
| 제품명 |
|
|||
|---|---|---|---|---|
| 성분 / 함량 |
|
|||
| 첨가제 | ||||
| 도핑금지 약물정보 |
|
|||
| 전문 / 일반 |
|
단일 / 복합 | ||
| 제조 / 수입사 | ||||
| 제형 | 투여경로 | |||
| 성상 | ||||
| 허가일 |
|
|||
| 의약품심사결과 | [변경] 타그리소정40밀리그램(오시머티닙메실산염) 외 1품목 의약품 품목변경허가 보고서(한국아스트라제네카(주) 타그리소정40밀리그램(오시머티닙메실산염) 외 1품목)(공개용)202501.pdf [변경] 타그리소정40밀리그램(오시머티닙메실산염) 외 1품목 의약품 품목변경허가 보고서(한국아스트라제네카(주) 타그리소정40밀리그램(오시머티닙메실산염) 외 1품목.pdf | |||
| 재심사 | ||||
| 대조 / 생동 | ||||
| 급여평가결과 | 급여(B), 2017년 제10차 타그리소정40,80밀리그램(아스트라제네카(주)).pdf | |||
| 급여정보 |
650700980 - 190108원/1정 급여(2026-01-01) - 190123원/1정 급여(2024-01-01) - 211248원/1정 급여(2022-03-01) - 217782원/1정 급여(2020-09-01) - 227312원/1정 급여(2020-01-01) - 227356원/1정 급여(2018-12-26) - 227356원/1정 급여(2017-12-05)
|
|||
| 급여인정기준 |
· [일반원칙] 국민건강보험 요양급여의 기준에 관한 규칙 제5조제4항에 의하여 중증환자 중 암환자에게 처방·투여하는 약제로서 건강보험심사평가원장이 정하여 공고하는 약제의 범위 및 비용부담 , 2025.05.01
|
|||
| ATC 코드 | ||||
| KPIC 약효분류 |
|
|||
| KPIC 학술 |
팜리뷰
약물 유발성 갑상샘 기능 이상(Drug-induced thyroid dysfunction), 약학정보원(대한약사회 지역의약품 안전센터), 2025-07-28
팜리뷰
[Special Issue] 암 환자의 퇴원 후 관리, 약학정보원(최은경), 2022-12-30
팜리뷰
항암치료와 구내염, 약학정보원(최선), 2017-06-12
팜리뷰
완화치료(palliative care), 약학정보원(남궁형욱), 2017-05-15
팜리뷰
항암화학요법 시 부작용 경감을 위한 한방의 병용(I) - 소화기계 부작용, 약학정보원(송보완), 2017-02-20
|
|||
| 대한약사저널 |
이슈트랜드
암 위험인자로서의 비만, 약학정보원 학술정보센터, 2020-08-10
|
|||
| 제품설명서 | 보 기 ( 2017-10-10 게시 ) | |||
| 의약품안전성 정보(DUR) |
||||
| 포장단위 (식약처 기준) |
||||
| 저장방법 | ||||
사용자GNB바
컨텐츠
의약품 상세정보

허가정보 ∙ 복약정보
효능 · 효과
단독요법
◦ EGFR 엑손 19 결손 또는 엑손 21 (L858R) 치환 변이가 있고 백금 ‑기반 화학 방사선 요법 중 또는 후 질병이 진행되지 않은 절제 불가능한 국소 진행성(III기) 비소세포폐암 환자의 치료
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 국소 진행성 또는 전이성 비소세포폐암 환자의 1차 치료
▪ 이전에 EGFR ‑TKI로 치료 받은 적이 있는 EGFR T790M 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자의 치료
병용요법
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 국소 진행성 또는 전이성 비편평 비소세포폐암 환자의 1차 치료에서 페메트렉시드와 백금 기반 항암화학요법과 병용 요법
용법 · 용량
이 약을 투여하는 경우, 치료 시작 전에 EGFR 변이 상태를 평가해야 한다. 다음에 대해 충분히 검증된 신뢰성 있는 시험방법을 사용하여 확인하여야 한다.
‑ 이전에 EGFR ‑TKI로 치료 받은 적이 있는 환자: T790M 변이; 식품의약품안전처에서 동 의약품의 사용에 적합하게 허가된 체외진단용 의료기기를 사용해야 함
단독요법
이 약의 권장 용량은 1일 1회 오시머티닙 80 mg이다.
병용요법
페메트렉시드와 백금 기반 항암화학요법과 병용할 때 이 약의 권장 용량은 1일 1회 오시머티닙 80 mg이다.
페메트렉시드와 시스플라틴 또는 카보플라틴 각각에 대한 투여량 정보는 해당 약물의 허가사항을 참조한다(13. 전문가를 위한 정보 참조).
이 약은 매일 일정한 시간에 식사와 관계없이 복용한다.
치료 기간
보조 치료 환자는 질병이 재발하거나 수용할 수 없는 독성이 나타날 때까지 복용을 지속한다. 3년을 초과하는 치료 기간은 연구되지 않았다.
국소 진행성 또는 전이성 폐암 환자는 질병이 진행되거나 허용할 수 없는 독성이 나타날 때까지 복용을 지속한다.
투여방법
이 약은 경구투여하며 물과 함께 통째로 삼켜야 한다. 정제를 부수거나 쪼개거나 씹어서는 안 된다.
환자가 정제를 삼킬 수 없는 경우에는, 우선 비탄산수 50 mL에 녹인다. 정제를 부수지 않고 물에 넣고 녹을 때까지 저은 후 즉시 마신다. 그 다음, 잔류물이 남지 않도록 물 약 100 mL을 추가하여 즉시 마신다. 다른 액체를 추가하지 않도록 한다.
비위관(nasogastric tube)을 통한 투여가 필요한 경우, 위와 동일한 과정을 따르되 처음 녹일 때 물 15 mL를 사용하고 잔류물을 헹구는데 15 mL를 사용한다.
약물을 녹인 용액과 잔류물을 헹군 용액은 정제를 물에 넣은 지 30분 안에 투여되어야 한다.
투여 누락
이 약의 복용을 누락한 경우, 다음 투여까지 12시간 이상 남았으면 즉시 복용한다.
용량 조절
각 환자의 안전성 및 내약성에 근거하여 투여 중단 및/또는 용량 감소가 필요할 수 있다. 용량 감소가 필요한 경우, 이 약의 용량은 1일 1회 40 mg으로 감량되어야 한다. 이상반응 독성에 대한 용량 감소 가이드라인은 표 1에 제시되어 있다.
표 1. 권장 용량 조절
|
표적 기관 |
이상반응a |
용량 변경 |
|
폐b |
간질성 폐질환(ILD)/폐염증 |
이 약을 영구 중단한다. |
|
확정적 백금-기반 화학 방사선 요법 후ILD/폐염증: 무증상(1등급) |
적절히 이 약을 지속하거나 일시 중지 및 재개한다. | |
|
확정적 백금-기반 화학 방사선 요법 후ILD/폐염증: 2등급 이상 |
이 약을 영구 중단한다. | |
|
심장b |
최소 2회의 별도의 ECG에서 500 msec 초과의 QTc 간격 |
QTc 간격이 481msec 미만이 될 때까지, 또는 베이스라인 QTc가 481msec 이상인 경우 베이스라인으로 회복될 때까지 이 약을 중단하고, 이후에 감량된 용량(40mg)으로 다시 시작한다. |
|
중대한 부정맥 징후/증상이 동반된 QTc 간격 연장 |
이 약을 영구 중단한다. | |
|
무증상성, 좌심실 박출률 기저치 대비 10 퍼센트포인트 이상 감소 및 50% 미만 |
최대 4주 동안 이 약 투여를 중지한다. 기저치로 돌아온다면 투여를 다시 시작한다. 기저치로 되돌아오지 않는다면 이 약을 영구 중단한다. | |
|
증상성 울혈성 심부전 |
이 약을 영구 중단한다. | |
|
피부b |
스티븐스-존슨증후군 및 독성 표피 괴사 용해 |
이 약을 영구 중단한다. |
|
혈액 및 림프계b |
재생 불량성 빈혈 |
이 약을 영구 중단한다. |
|
기타 |
3등급 이상의 이상반응 |
최대 3주간 이 약을 중단한다. |
|
3등급 이상의 이상반응이 이 약을 최대 3주간 중단한 이후 0-2등급으로 개선되는 경우 |
이 약을 동일 용량(80 mg) 또는 저용량(40 mg)으로 다시 시작 할 수 있다. | |
|
최대 3주간 이 약을 중단한 이후 0-2등급으로 개선되지 않는 3등급 이상의 이상반응 |
이 약을 영구 중단한다. |
b 사용상의 주의사항 1. 경고 참조.
병용요법: 이 약을 페메트렉시드 및 백금 기반 항암화학요법과 병용투여 시 이상반응이 발생한 경우, 투여 약물 중 어느 하나의 투여를 적절히 조절해야 한다. 이 약의 용량 조절은 표 1을 참고한다. 페메트렉시드, 시스플라틴 또는 카보플라틴은 각각의 허가사항에 따라 투여 중지, 용량 감소, 또는 영구 중단해야 한다.
사용상의 주의사항
1. 경고
ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들에서 이 약을 단독요법으로 투여받은 1813명의 환자 중 간질성 폐질환 또는 간질성 폐질환 유사 이상반응(예. 폐염증)은 4.0%의 환자에게서 보고되었으며, 이 중 치명적인 건은 0.4%(n=7) 보고되었다. 간질성 폐질환은 일본인 환자의 11.2%, 일본인이 아닌 아시아인 환자의 2.3%, 비아시아인 환자의 2.7%에게서 발생하였다. 첫 번째 투여부터 간질성 폐질환 또는 간질성 폐질환 유사 이상반응들의 발현시점의 중앙값은 2.8개월이었다.
FLAURA2에서 페메트렉시드 및 백금 기반 항암화학요법과 병용하여 이 약을 투여받은 276명의 환자 중 간질성 폐질환 또는 간질성 폐질환 유사 이상반응은 3.3%에서 보고되었으며, 이 중 치명적인 건은 0.4% (n=1)이었다. 간질성 폐질환은 일본인 환자의 14.9% 및 비아시아인 환자의 1.7%에게서 발생하였다. FLAURA2 병용요법 군에서 일본인이 아닌 아시아인 환자에서는 간질성 폐질환이 발생하지 않았다. 첫 번째 투여부터 간질성 폐질환 또는 간질성 폐질환 유사 이상반응들의 발현시점의 중앙값은 5.3개월이었다.
간질성 폐질환을 시사할 수 있는 호흡기 증상(예: 호흡곤란, 기침, 발열)의 악화가 나타나는 환자는 이 약을 중단하고 즉시 간질성 폐질환에 대해 조사한다. 간질성 폐질환이 확인되면 이 약을 영구 중단한다.
확정적 백금‑기반 화학 방사선 요법 후 간질성 폐질환
LAURA 임상시험에서 확정적 백금‑기반 화학 방사선 요법 후, 이 약을 투여받은 143명의 환자 중 56%(80명)와 위약을 투여받은 73명의 환자 중 38%(28명)에서 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응(예: 폐염증)이 보고되었다. 이 약 군에서는 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응 중 0.7%(1명)의 치명적 증례가 보고되었으며, 3등급은 3.5%, 2등급은 34%, 1등급은 18%이었다. 이 약을 투여받은 환자 중 7%가 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응으로 투여를 영구 중단 했으며, 35%는 일시 중단했다. 재투여를 받은 환자 46명 중 11%는 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 재발하였다. 이 약을 투여받고 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생한 80명의 환자 중 40%는 회복, 1.3%는 후유증을 동반하고 회복, 16%는 회복 중이었으며, 41%는 회복되지 않고 1.3%(1명)는 사망하였다.
확정적 백금‑기반 화학 방사선 요법 후 이 약으로 치료받은 환자에서 간질성 폐질환을 시사할 수 있는 호흡기 증상(예: 호흡곤란, 기침, 발열)의 악화가 나타나는 환자는 이 약을 중단하고 즉시 간질성 폐질환에 대해 조사한다. 간질성 폐질환이 확인되면 무증상(1등급) 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생하는 경우, 적절히 이 약을 지속하거나 일시 중지 및 재개한다. 2등급 이상의 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생하는 경우, 이 약의 사용을 영구 중단한다.
2) 스티븐스‑존슨증후군, 다형성 홍반 (Erythema multiforme) 및 독성 표피 괴사 용해
이 약 치료와 관련하여, 다형성 홍반 및 독성 표피 괴사 용해의 사례 보고는 흔하지 않게 보고되었고, 스티븐스‑존슨증후군은 드물게 보고되었다. 치료를 시작하기 전에 환자에게 스티븐스‑존슨증후군, 다형성 홍반 및 독성 표피 괴사 용해의 징후와 증상을 알려야 한다.
스티븐스‑존슨증후군 또는 독성 표피 괴사 용해를 연상시키는 징후와 증상이 나타나면, 이 약을 일시 중단해야 한다. 스티븐스‑존슨증후군 또는 독성 표피 괴사 용해가 진단되면, 이 약을 즉시 중지해야 한다.
다형성 홍반을 연상시키는 징후와 증상이 있는 경우, 긴밀한 환자 모니터링 및 이 약의 중단 또는 중지를 고려해야 한다.
3) QTc 간격 연장
이 약 단독요법 (80mg)으로 치료한 ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들의 1813명 환자 중, 1.1%(n=20)는 500 msec 초과 QTc를, 4.3%(n=78)는 베이스라인 QTc로부터 60 msec를 초과하는 QTc 증가를 나타냈다. 이 약에 대한 약동학적 분석 시 QTc 간격 연장의 농도 의존적 증가가 예측되었다. ADAURA, LAURA, FLAURA, FLAURA2 또는 AURA 임상시험들에서 QTc와 관련된 부정맥 사례는 보고되지 않았다.
가능한 경우, 선천적으로 긴 QT 증후군이 있는 환자는 이 약의 사용을 피한다 (3.이상반응 참조). 울혈성 심부전, 전해질 이상이 있는 환자, 또는 QTc 간격이 연장되는 것으로 알려진 약물을 투여 중인 환자에서는 심전도(ECG) 및 전해질의 주기적인 모니터링을 고려한다. 심전도 검사에서 QTc 간격이 500 msec을 초과하는 결과가 적어도 2회 별도로 발생한 환자에서는 QTc 간격이 481msec 미만이 될 때까지, 또는 베이스라인 QTc가 481msec 이상인 경우 베이스라인으로 회복될 때까지 이 약을 중단하고, 이후에 용법용량 항 표 1에 기술된 바와 같이 감소된 용량으로 투여를 재개한다. 다음 중 어느 하나와 더불어 QTc 간격 연장이 발생한 환자는 이 약을 영구 중단한다: 염전성 심실빈맥(Torsade de pointes), 다형성 심실빈맥(polymorphic ventricular tachycardia), 중대한 부정맥의 징후/증상.
4) 심수축성/심장독성 변화
임상시험들에서 이 약을 투여받은 1813명의 환자 중 3.8%에서 심장독성(심부전, 만성 심부전, 울혈성 심부전, 폐부종, 박출률 감소)을 나타냈다. 이 중 1명(0.1%)은 치명적인 결과로 이어졌다.
임상시험에서 베이스라인 및 최소 1회의 좌심실박출률 추적 평가를 받은 이 약의 단독요법으로 치료받은 환자의 4.2%(65/1557)에서 좌심실 박출량이 10퍼센트포인트 이상 감소하고 50% 미만으로 나타났다. 위약 대조 시험(ADAURA)에서 이 약을 투여 받은 환자의 1.5%(5/325)와 위약을 투여 받은 환자의 1.5%(5/331)에서 10%포인트 이상 50% 미만의 좌심실박출률 감소를 경험했다. LAURA 임상시험에서 백금‑기반 화학 방사선 요법 후, 이 약으로 치료받은 환자의 3.0% (4/135명)와 위약으로 치료받은 환자 0명에서 좌심실 박출량이 10퍼센트포인트 이상 감소하고 50% 미만으로 나타났다. FLAURA2에서 베이스라인 및 최소 1회의 좌심실박출률 추적 평가를 받은 이 약과 페메트렉시드, 백금 기반 항암화학요법을 병용하여 치료받은 환자의 8.0%(21/262)에서 좌심실 박출량이 10%포인트 이상 감소하고 50% 미만으로 나타났다.
심장 위험인자가 있거나 좌심실박출률에 영향을 미칠 수 있는 상태의 환자에게는 베이스라인 및 치료 중 좌심실박출률 평가를 포함하는 심장 모니터링을 한다. 치료 중 관련된 심장 징후/증상이 있는 환자의 경우, 좌심실박출률 평가를 포함한 심장 모니터링을 한다. 좌심실박출률이 기저치 대비 10 퍼센트포인트 이상 감소하고 50% 미만으로 나타날 경우, 이 약 투여를 최대 4주간 중단하고 중단 후에도 증상이 개선되지 않을 때에는 이 약 투여를 영구 중단한다. 증상성 울혈성 심부전 발생 시에는 이 약의 투여를 영구 중단한다.
5) 각막염
ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들에서 이 약의 단독요법으로 치료된 1813명의 환자 중 0.6%(n=10)에서 각막염이 보고되었다. 다음과 같은 각막염의 징후 및 증상을 보이는 환자들은 즉시 안과전문의에게 상담을 받아야 한다: 눈의 염증, 눈물 분비, 빛에 민감한 눈, 시야 흐림, 눈의 통증 그리고/또는 눈의 충혈 (용법용량 항 참조)
6) 재생 불량성 빈혈
재생 불량성 빈혈이 이 약의 치료와 관련하여 드물게 보고되었다. 일부 사례는 치명적인 결과가 있었다. 치료를 시작하기 전에 환자에게 지속적인 발열, 멍, 출혈, 창백을 포함하되 이에 국한되지 않는 재생 불량성 빈혈의 징후와 증상에 대해 알려야 한다. 재생 불량성 빈혈을 암시하는 징후 및 증상이 발생하면 환자를 면밀히 모니터링하고 이 약의 중지 또는 중단을 고려해야 한다. 재생 불량성 빈혈이 확인된 환자는 이 약을 중단해야 한다.
2. 다음 환자에는 투여하지 말 것.
이 약의 주성분이나 부형제에 대해 중증의 과민증이 있는 환자
3. 이상반응
1) 안전성 프로파일의 전반적인 요약
EGFR 변이 양성 비소세포폐암 환자에 대한 시험
이 약 단독요법의 안전성은 EGFR 변이 양성 비소세포폐암 환자 1813명으로부터 수집된 데이터를 기반으로 한다. 이러한 환자들은 4건의 무작위배정된 3상 임상시험(ADAURA: 보조 치료; FLAURA 및 FLAURA2(단독요법 군):1차 치료; AURA3: 2차 치료), 2건의 단일군 2상 임상시험(AURAex 및 AURA2: 2차 치료 이상) 및 1건의 1상 임상시험(AURA1: 1차 치료 이상)에서 이 약 1일 80 mg을 투여 받았다.
대부분의 이상반응 중증도는 1등급 또는 2등급이었다. 가장 흔한 빈도로 보고된 약물이상반응은 설사(47%), 발진(46%), 손발톱주위염(34%), 건성 피부(32%), 및 구내염(24%)이었다. 이 약에서 나타난 3등급 및 4등급 이상반응은 각각 9.2%와 0.2%였다. 이 약 80 mg을 1일 1회 치료받은 환자들에게서, 약물이상반응으로 인한 용량감소는 3.6%의 환자들에게서 나타났다. 이상반응으로 인한 투약중단은 4.7%이었다.
백금‑기반 화학 방사선 요법 후 이 약(1일 1회 80 mg)의 안전성은 EGFR 돌연변이 양성 비소세포폐암 환자 143명에서 얻은 자료를 기반으로 한다.
페메트렉시드와 백금 기반 항암화학요법을 병용하여 투여한 이 약의 안전성은 EGFR 변이 양성 비소세포폐암 환자 276명 데이터를 기반으로 하며, 이 약의 단독요법 및 페멕트렉시드와 백금 기반 항암화학요법의 알려진 안전성 프로파일과 일치했다.
스테로이드 치료가 필요한 간질성 폐질환, 약물 유도 간질성 폐질환, 방사선 폐염증의 과거 병력이 있거나 임상적으로 활동성 간질성 폐질환 증거가 있는 환자는 임상시험에서 제외되었다. 안정 시 ECG (예를 들어, QTc 간격이 470 ms 초과)로 측정 시 리듬 및 전도가 임상적으로 중요한 비정상인 환자는 이러한 시험들에서 제외되었다. 스크리닝 및 이후 12주마다 환자의 좌심실박출률이 평가되었다.
AURAex 및 AURA2에 참여한 한국인 환자(66명)에서 전체 및 3/4등급 이상반응 발생율은 전체 환자군과 유사하였다. 다만, 한국인 환자에서 전체 환자군에 비해 상대적으로 많이 발생하는 이상반응은 발진, 소양증, 손발톱주위염, 혈소판 감소증, 백혈구 감소증으로 나타났다. 상대적으로 낮은 빈도로 발생하는 이상반응은 설사, 오심으로 보고되었다. 이들 이상반응은 대부분 1등급 또는 2등급이었다.
2) 이상반응 표
이상반응은 ADAURA, FLAURA, FLAURA2, AURA3, AURAex, AURA2, 및 AURA1 시험들에서 이 약의 단독요법으로 1일 80 mg을 투여 받은 EGFR 변이 양성 환자 1813명의 통합 데이터세트에서의 비교 가능한 이상반응 발생률에 근거하여 표 2에 빈도 카테고리로 분류되었다.
약물이상반응은 MedDRA 기관계 분류(System Organ Class;SOC)에 따라 나열하였다. 각 기관계 분류 내에서, 약물이상반응은 빈도 내림차순으로(즉, 가장 빈번한 반응이 제일 먼저 오도록) 나열하였다. 각 빈도 군 내에서, 약물이상반응은 중대성이 감소하는 순으로 나열하였다. 또한, 각 약물이상반응의 해당 빈도 범주는 CIOMS III 분류에 근거하여 다음과 같이 정의한다: 매우 흔하게(≧1/10), 흔하게(≧1/100 ~ <1/10), 흔하지 않게(≧1/1,000 ~ <1/100), 드물게(≧1/10,000 ~ <1/1000), 매우 드물게(<1/10,000), 빈도불명(사용 가능한 데이터로부터 추정할 수 없음).
표 2. ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 시험들에서 보고된 이상반응a
|
MedDRA SOC |
CIOMS 분류/전체 빈도(모든 CTCAE 등급b) |
CTCAE 3등급 이상의 빈도 |
|
MedDRA 선호 용어 | ||
|
혈액 및 림프계 이상 | ||
|
재생 불량성 빈혈 |
드물게(0.06%) |
0.06% |
|
눈 이상 | ||
|
각막염c |
흔하지 않게(0.6%) |
0.06% |
|
호흡계, 흉부 및 종격 이상 | ||
|
비출혈 |
흔하게(6%) |
0% |
|
간질성 폐질환d |
흔하게(4.0%)e |
1.4% |
|
위장관계 이상 | ||
|
설사 |
매우 흔하게(47%) |
1.4% |
|
구내염f |
매우 흔하게(24%) |
0.4% |
|
피부 및 피하조직 이상 | ||
|
발진g |
매우 흔하게 (46%) |
0.8% |
|
손발톱주위염h |
매우 흔하게 (34%) |
0.4% |
|
건성 피부i |
매우 흔하게 (32%) |
0.1% |
|
소양증j |
매우 흔하게 (17%) |
0.06% |
|
탈모증 |
흔하게(5%) |
0% |
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
흔하게(2.1%) |
0% |
|
두드러기 |
흔하게(1.9%) |
0.1% |
|
피부 과다 색소 침착k |
흔하게(1.0%) |
0% |
|
다형성 홍반l |
흔하지 않게(0.3%) |
0% |
|
독성 표피 괴사 용해m |
흔하지 않게(0.2%) | |
|
피부 혈관염m |
흔하지 않게(0.2%) | |
|
스티븐스-존슨증후군n |
드물게(0.02%) | |
|
실험실적 수치 | ||
|
혈액 크레아틴 인산 활성 효소 증가 |
흔하게(1.9%) |
0.3% |
|
QTc 간격 연장o |
흔하게(1.1%) | |
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||
|
백혈구 감소p |
매우 흔하게(65%) |
1.8% |
|
림프구 감소p |
매우 흔하게(64%) |
8% |
|
혈소판 수 감소p |
매우 흔하게(53%) |
1.3% |
|
호중구 감소p |
매우 흔하게(36%) |
4.0% |
|
혈액 크레아티닌 증가p |
흔하게(9%) |
0.2% |
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란, 각막내피결손, 각막염, 점상각막염 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질성 폐렴 포함.
e 7 건의 CTCAE 5등급(치명적)이 보고됨.
f 입 궤양 형성, 구내염 포함
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성 발진(rash vesicular), 피부미란(skin erosion) 포함.
h 손발톱바닥장애(nail bed disorder), 손발톱바닥감염(nail bed infection), 손발톱바닥염증(nail bed inflammation), 손발톱변색(nail discolouration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k 시판 후 조사에서 지속색소이상홍반 사례가 보고되었다.
l ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 시험의 1813명 환자 중 6명이 다형성 홍반을 보고하였다. 시판 후 조사 연구(N=3578)로부터의 7건을 포함하여, 다형성 홍반의 시판 후 보고도 있었다.
m 추정 빈도. 추정치에 대한 95% CI의 상한은 3/1813 (0.17%)이다. 임상시험에서 보고 없음.
n 시판 후 연구에서 보고된 한 건의 사례이며, 빈도는 ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 연구와 시판 후 연구(N=5391)에서 도출되었다.
o QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
p 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 3. ADAURAa시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=337) |
위약 (N=343) | ||||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%)c |
모든 등급 (%) |
3등급 이상 (%)c | ||
|
MedDRA 선호 용어 | ||||||
|
눈 이상 | ||||||
|
각막염d |
0.6 |
0 |
0.3 |
0 | ||
|
호흡계, 흉부 및 종격 이상 | ||||||
|
비출혈 |
5.6 |
0 |
0.9 |
0 | ||
|
간질성 폐질환e |
3.0 |
0 |
0 |
0 | ||
|
위장관계 이상 | ||||||
|
설사 |
46.3 |
2.4 |
19.8 |
0.3 | ||
|
구내염f |
28.2 |
1.8 |
6.4 |
0 | ||
|
피부 및 피하조직 이상 | ||||||
|
발진g |
39.2 |
0.3 |
19.0 |
0 | ||
|
손발톱주위염h |
36.5 |
0.9 |
3.8 |
0 | ||
|
건성 피부i |
29.4 |
0.3 |
7.3 |
0 | ||
|
소양증j |
19.3 |
0 |
8.7 |
0 | ||
|
탈모증 |
5.6 |
0 |
2.0 |
0 | ||
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
1.8 |
0 |
0 |
0 | ||
|
피부 과다 색소 침착 |
1.8 |
0 |
0 |
0 | ||
|
두드러기 |
1.5 |
0 |
0.3 |
0.3 | ||
|
실험실적 수치 | ||||||
|
혈액 크레아틴 인산 활성 효소 증가 |
3.3 |
0.9 | ||||
|
QTc 간격 연장k |
0.6 |
0 | ||||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||||||
|
백혈구 감소l |
54.0 |
0 |
25.4 |
0 | ||
|
혈소판 수 감소l |
47.2 |
0 |
6.6 |
0.3 | ||
|
림프구 감소l |
43.8 |
2.2 |
14.4 |
0.9 | ||
|
호중구 감소l |
25.6 |
0.3 |
10.2 |
0.3 | ||
|
혈액 크레아티닌 증가l |
9.8 |
0 |
4.5 |
0.3 | ||
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0
c 모든 사례는 3등급이었다. 사망은 없었다.
d 각막염(keratitis), 점상각막염(punctate keratitis), 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect) 포함.
e 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
f 구내염(stomatitis), 입 궤양 형성(mouth ulceration) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis), 전신 소양증(pruritis generalised) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 4. LAURAa 시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=143) |
위약 (N=73) | ||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
MedDRA 선호 용어 | ||||
|
눈 이상 | ||||
|
각막염c |
0.7 |
0 |
1.4 |
0 |
|
호흡계, 흉부 및 종격 이상 | ||||
|
간질성 폐질환d |
8 |
2.1e |
1.4 |
0 |
|
비출혈 |
0.7 |
0 |
0 |
0 |
|
위장관계 이상 | ||||
|
설사e |
36 |
2.1 |
14 |
0 |
|
구내염f |
15 |
0 |
4.1 |
0 |
|
피부 및 피하조직 이상 | ||||
|
발진g |
36 |
0.7 |
19 |
0 |
|
손발톱 주위염h |
23 |
0 |
1.4 |
0 |
|
건성 피부i |
17 |
0.7 |
5 |
0 |
|
소양증j |
13 |
0 |
7 |
0 |
|
탈모증 |
1.4 |
0 |
0 |
0 |
|
두드러기 |
1.4 |
0 |
1.4 |
0 |
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
0 |
0 |
0 |
0 |
|
피부 과다 색소 침착 |
0 |
0 |
0 |
0 |
|
다형성 홍반 |
0 |
0 |
0 |
0 |
|
실험실적 수치 | ||||
|
혈액 크레아틴 인산 활성 효소 증가 |
3.5 |
1.4 |
0 |
0 |
|
QTc 간격 연장k |
0.7 |
0 | ||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||||
|
림프구 감소l |
70 |
3.5 |
40 |
1.4 |
|
백혈구 감소l |
66 |
2.8 |
24 |
0 |
|
혈소판 수 감소l |
51 |
1.4 |
8 |
1.4 |
|
중성구 감소l |
42 |
2.1 |
15 |
1.4 |
|
혈액 크레아티닌 증가l,m |
19 |
0 |
12 |
0 |
a 무작위 배정된 치료로서 1회 용량 이상 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란, 각막내피결손, 각막염, 점상각막염 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질성 폐렴 포함.
e 1건의 CTCAE 5등급(치명적)이 보고됨.
f 입 궤양, 구내염 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성 발진(rash vesicular), 피부미란(skin erosion) 포함.
h 손발톱바닥장애(nail bed disorder), 손발톱바닥감염(nail bed infection), 손발톱바닥염증(nail bed inflammation), 손발톱변색(nail discolouration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부 건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k QTcF 연장이 >500 msec를 보인 환자의 발생률을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
m LAURA 시험의 베이스라인 혈액 크레아티닌 청소율(<30 mL/min)은 이 약의 다른 단독요법 시험들에서의 베이스라인 혈액 크레아틴 청소율(<50 mL/min)보다 낮으므로, 등급 변화가 일어날 가능성이 더 높았다.
표 5. FLAURAa 시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=279) |
EGFR TKI 대조약 (게피티닙 또는 엘로티닙) (N=277) | ||||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) | ||
|
MedDRA 선호 용어 | ||||||
|
눈 이상 | ||||||
|
각막염c |
0.4 |
0 |
1.4 |
0 | ||
|
호흡계, 흉부 및 종격 이상 | ||||||
|
비출혈 |
6.1 |
0 |
5.1 |
0 | ||
|
간질성 폐질환d |
3.9 |
1.1 |
2.2 |
1.4 | ||
|
위장관계 이상 | ||||||
|
설사e |
58 |
2.2 |
57 |
2.5 | ||
|
구내염f |
32 |
0.7 |
22 |
1.1 | ||
|
피부 및 피하조직 이상 | ||||||
|
발진g |
58 |
1.1 |
78 |
6.9 | ||
|
건성 피부h |
36 |
0.4 |
36 |
1.1 | ||
|
손발톱주위염i |
35 |
0.4 |
33 |
0.7 | ||
|
소양증j |
17 |
0.4 |
17 |
0 | ||
|
탈모증 |
7.2 |
0 |
13 |
0 | ||
|
두드러기 |
2.2 |
0.7 |
0.4 |
0 | ||
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
1.4 |
0 |
2.5 |
0 | ||
|
피부 과다 색소 침착 |
0.4 |
0 |
1.1 |
0 | ||
|
실험실적 수치 | ||||||
|
QTc 간격 연장k |
1.1 |
0.7 | ||||
|
혈액 크레아틴 인산 활성 효소 증가 |
0.4 |
0.4 | ||||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||||||
|
백혈구 감소l |
72 |
0.4 |
31 |
0.4 | ||
|
림프구 감소l |
63 |
5.6 |
36 |
4.2 | ||
|
혈소판수 감소l |
51 |
0.7 |
12 |
0.4 | ||
|
호중구 감소l |
41 |
3.0 |
10 |
0 | ||
|
혈액 크레아티닌 증가l |
8.8 |
0 |
6.7 |
0.4 | ||
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0.
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
e EGFR TKI 대조약 군에서 1건의 CTCAE 5등급(치명적) 사건이 보고되었다.
f 입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
i손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis), 전신 소양증(pruritis generalised) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
l실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 6. FLAURA2a 시험에서 보고된 이상반응
|
MedDRA SOC |
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=276) |
이 약 (N=275) | |||||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) | |||
|
MedDRA 선호 용어 | |||||||
|
눈 이상 | |||||||
|
각막염c |
0.7 |
0 |
0 |
0 | |||
|
호흡계, 흉부 및 종격 이상 | |||||||
|
비출혈 |
7 |
0.4 |
7 |
0 | |||
|
간질성 폐질환d |
3.3 |
0.7e |
3.6 |
1.8e | |||
|
위장관계 이상 | |||||||
|
설사 |
43 |
2.9 |
41 |
0.4 | |||
|
구내염f |
31 |
0.4 |
21 |
0.4 | |||
|
피부 및 피하조직 이상 | |||||||
|
발진g |
49 |
2.5 |
44 |
1.5 | |||
|
손발톱주위염h |
27 |
0.7 |
32 |
0.4 | |||
|
건성 피부i |
24 |
0 |
31 |
0 | |||
|
탈모증 |
9 |
0 |
5 |
0 | |||
|
소양증j |
8 |
0 |
11 |
0 | |||
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
5 |
0 |
3.3 |
0 | |||
|
피부 과다 색소 침착 |
2.5 |
0 |
1.1 |
0 | |||
|
두드러기 |
1.4 |
0.4 |
1.5 |
0 | |||
|
다형성 홍반 |
1.4 |
0.7 |
0.4 |
0 | |||
|
실험실적 수치 | |||||||
|
혈액 크레아틴 인산 활성 효소 증가 |
3.3 |
1.1 |
3.3 |
0 | |||
|
QTc 간격 연장k |
1.8 |
1.8 | |||||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | |||||||
|
백혈구 감소l |
88 |
20 |
53 |
3.3 | |||
|
혈소판수 감소l |
85 |
16 |
44 |
1.8 | |||
|
호중구 감소l |
85 |
36 |
40 |
4.7 | |||
|
림프구 감소l |
78 |
16 |
55 |
7 | |||
|
혈액 크레아티닌 증가l |
22 |
0.4 |
8 |
0 | |||
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질화 폐렴(organising pnuemonia) 포함.
e 1건의 CTCAE 5등급(치명적) 사건이 보고되었다.
f 입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular) 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
I 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 7. AURA3a시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=279) |
항암화학요법 (페메트렉시드/시스플라틴 또는 페메트렉시드/카보플라틴) (N=136) | ||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
MedDRA 선호용어 | ||||
|
눈 이상 | ||||
|
결막염c |
1.1 |
0 |
0.7 |
0 |
|
호흡계, 흉부 및 종격 이상 | ||||
|
비출혈 |
5.4 |
0 |
1.5 |
0 |
|
간질성 폐질환d,e |
3.6 |
0.4 |
0.7 |
0.7 |
|
위장관계 질환 | ||||
|
설사 |
41 |
1.1 |
11 |
1.5 |
|
구내염f |
19 |
0 |
15 |
1.5 |
|
피부 및 피하조직 질환 | ||||
|
발진g |
34 |
0.7 |
5.9 |
0 |
|
건성 피부h |
23 |
0 |
4.4 |
0 |
|
손발톱주위염i |
22 |
0 |
1.5 |
0 |
|
소양증j |
13 |
0 |
5.1 |
0 |
|
탈모증 |
3.6 |
0 |
2.9 |
0 |
|
두드러기 |
2.5 |
0 |
1.5 |
0 |
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
1.8 |
0 |
0.7 |
0 |
|
피부 과다 색소 침착 |
0.4 |
0 |
3.7 |
0 |
|
실험실적 수치 | ||||
|
QTc 간격연장k |
1.4 |
0 | ||
|
혈액 크레아틴 인산 활성 효소 증가 |
0.7 |
0.7 | ||
|
(CTCAE grade 이동으로 나타난 시험 결과를 근거로 한 결과) | ||||
|
백혈구 감소l |
61 |
1.1 |
75 |
5.3 |
|
혈소판수 감소l |
46 |
0.7 |
48 |
7.4 |
|
호중구 감소l |
27 |
2.2 |
49 |
12 |
|
혈액 크레아티닌 증가l |
6.5 |
0 |
9.2 |
0 |
a 최소 1회 투여를 한 환자의 사례만 요약되어 있다.
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
e CTCAE 5등급(치명적) 1건이 보고되었다.
f입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 농포성 발진(rash pustular) 포함.
h 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 건조증(xerosis) 포함.
i손발톱바닥장애(nail bed disorders), 손발톱바닥염증(nail bed inflammation), 손발톱바닥압통(nail bed tenderness), 손발톱변색(nail discoloration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱능성형성(nail ridging), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱주위염(paronychia) 포함.
j 눈꺼풀 소양증, 소양증, 전신 소양증 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
단일군 임상2상 AURAex 및 AURA2 임상시험에서의 안전성 결과는 일반적으로 AURA3의 이 약 군에서 관찰된 것과 일치하였다. 추가적이거나 예상하지 못한 독성이 관찰되지 않았으며, 이상반응은 종류, 중증도 및 빈도 등이 알려진 것과 유사하였다.
선택된 이상반응에 대한 설명
혈액학적 독성
이 약으로 치료를 받은 환자에서 실험실적 백혈구, 림프구, 호중구 및 혈소판 수의 중앙값에 조기 감소가 관찰되었으며, 이는 시간이 지남에 따라 안정해진 후 정상 하한보다 높게 유지되었다. 백혈구 감소증, 림프구 감소증, 호중구 감소증, 및 혈소판 감소증 이상사례가 보고되었으며, 대부분은 경증 또는 중등증이었고 투여 중지로 이어지지 않았다.
3) 국내 시판 후 수집된 중대한 이상사례 분석▪평가 결과 확인된 이상사례는 다음과 같다. 다만, 이로써 곧 해당성분과 다음의 이상사례 간에 인과관계가 입증된 것을 의미하는 것은 아니다.
◦ 호흡기계 : 폐색전증
※ 국내 시판 후 조사 결과
국내에서 1178명을 대상으로 실시한 시판 후 조사 결과, 이상사례의 발현율은 인과관계와 상관없이 69.10%(814/1178명, 2287건)로 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응 및 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 발현 빈도에 따라 아래 표에 나열하였다.
|
발현빈도 |
기관계 |
중대한 약물이상반응 6.79% (80/1178명, 99건) |
예상하지 못한 약물이상반응 17.74% (209/1178명, 296건) |
|
흔하게 (1~10% 미만) |
임상 검사 |
- |
알라닌 아미노 전이 효소 증가, 아스파르트산 아미노 전이 효소 증가 |
|
각종 위장관 장애 |
- |
변비 | |
|
대사 및 영양 장애 |
- |
식욕 감소 | |
|
전신 장애 및 투여 부위 병태 |
- |
점막 염증 | |
|
흔하지 않게 (0.1~1% 미만) |
호흡기, 흉곽 및 종격 장애 |
폐염증, 간질성 폐 질환, 폐 색전증 |
호흡 곤란, 기침, 흉막 삼출 |
|
혈액 및 림프계 장애 |
중성구 감소증, 혈소판 감소증 |
빈혈 | |
|
각종 위장관 장애 |
구역, 설사, 복통 |
구토, 소화 불량, 복통, 상복부 통증, 입 건조, 식도염, 위염 | |
|
감염 및 기생충 감염 |
폐렴 |
폐렴 | |
|
신장 및 요로 장애 |
급성 신 손상 |
급성 신 손상 | |
|
피부 및 피하 조직 장애 |
약물 발진 |
피부 병변, 피부 탈락, 피부 반응 | |
|
전신 장애 및 투여 부위 병태 |
통증 |
무력증, 피로, 말초 부종, 통증, 전신 부종 | |
|
간담도 장애 |
간염, 약물-유발 간 손상 |
간염, 약물-유발 간 손상 | |
|
대사 및 영양 장애 |
- |
섭식 저하 | |
|
각종 신경계 장애 |
- |
말초 신경 병증, 감각 저하, 어지러움, 두통, 지각 이상 | |
|
각종 심장 장애 |
- |
심장막 삼출 | |
|
근골격 및 결합 조직 장애 |
- |
관절통 | |
|
각종 눈 장애 |
- |
눈 건조 | |
|
드물게 (0.01~0.1% 미만) |
호흡기, 흉곽 및 종격 장애 |
호흡 곤란, 흉막 삼출 |
구인두 통증, 폐 침윤 |
|
혈액 및 림프계 장애 |
발열성 중성구 감소증, 백혈구 감소증 |
발열성 중성구 감소증, 백혈구증 | |
|
각종 위장관 장애 |
구토, 흑색변 |
위 식도 역류 질환, 위 궤양, 흑색변, 혀 발진, 혀 궤양 형성 | |
|
감염 및 기생충 감염 |
요로 감염, 충수염, 클로스트리듐 디피실레 결장염, 위장염 |
요로 감염, 위장염, 구강 칸디다증, 부비동염, 충수염, 클로스트리듐 디피실레 결장염, 림프선 감염 | |
|
각종 심장 장애 |
급성 심근 경색, 심부전, 심근 병증, 심독성, 확장 심근 병증, 두근거림, 심장막 삼출 |
심부전, 두근거림, 급성 심근 경색, 심근 병증, 심독성, 확장 심근 병증, 심실 위 빈맥 | |
|
신장 및 요로 장애 |
만성 신장병, 신부전, 신 기능 장애 |
야간뇨, 만성 신장병, 신부전, 신 기능 장애 | |
|
피부 및 피하 조직 장애 |
피부 혈관염, 피부염, 발진 |
수포, 모낭 장애, 손발톱 소피 균열, 건선, 딱지, 피부 삼출 | |
|
각종 신경계 장애 |
어지러움, 뇌 병증, 말초 신경 병증, 신경 독성 |
진전, 당뇨 신경 병증, 신경통, 말초 감각 신경 병증, 뇌 병증, 신경 독성 | |
|
전신 장애 및 투여 부위 병태 |
무력증, 비-심장성 흉통 |
발열, 안면 부종, 부종, 국소 부종, 비-심장성 흉통 | |
|
대사 및 영양 장애 |
식욕 감소, 당뇨병, 고칼슘 혈증 |
당뇨병, 고칼슘 혈증 | |
|
양성, 악성 및 상세 불명의 신생물(낭종 및 용종 포함) |
위장관 선종, 악성 흉막삼출액 |
위장관 선종, 악성 흉막삼출액 | |
|
임상 검사 |
심전도 QT 연장, 중성구 수 감소 |
혈액 알칼리 인산 분해 효소 증가, 간 효소 증가 | |
|
각종 혈관 장애 |
말초 동맥 협착 |
열감 홍종, 말초 동맥 협착 | |
|
각종 내분비 장애 |
부신 부전 |
부신 부전 | |
|
근골격 및 결합 조직 장애 |
- |
근육통, 근육 쇠약, 옆구리 통증, 관절 주위염, 근육 연축, 관절염 | |
|
각종 눈 장애 |
- |
백내장, 시각 장애, 시야 흐림, 눈 분비물 | |
|
생식계 및 유방 장애 |
- |
질 출혈, 자궁 경부 용종, 월경 사이 출혈 |
각종 신경계 장애 ‑ 미각 이상
근골격 및 결합 조직 장애 ‑ 등허리 통증, 사지 통증, 근육 연축
대사 및 영양 장애 ‑ 식욕 감소
전신 장애 및 투여 부위 병태 ‑ 무력증, 피로
4. 일반적 주의
1) EGFR 변이 상태 및 검사
비소세포폐암 환자에서 완전 종양 절제술 후 보조 치료로서 이 약의 사용을 고려할 때, EGFR 변이 양성 상태 (엑손 19 결손 또는 엑손 21[L858R] 치환 변이)는 치료 적격성을 나타낸다.
이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
절제 불가능한 국소 진행성(III기) 비소세포폐암 환자에서 이 약의 사용을 고려할 때, EGFR 돌연변이 양성 상태(엑손 19 결손 또는 엑손 21 [L858R] 치환 변이)는 치료 적격성을 나타낸다. 이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
국소 진행성 또는 전이성 비소세포폐암의 치료제로서 이 약의 사용을 고려할 때, 이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
단독요법에서 보조 치료 임상시험에서는 EGFR 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였다.
단독요법 및 페메트렉시드와 백금 기반 항암화학요법과 병용 요법의 1차 치료 임상시험에서는 EGFR 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였고, 인증받은 지역 실험실 검사도 사용되었다.
임상시험에서는 T790M 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였다.
2) 이 약은 운전 및 기계 조작하는 능력에 영향을 미치지 않거나 또는 현저한 영향을 미치지 않는다.
5. 상호작용
1) 이 약의 혈장 농도를 증가시킬 수 있는 활성 물질
생체 외(in vitro) 시험에서 이 약의 1단계 대사는 주로 CYP3A4 및 CYP3A5를 통해 일어나는 것으로 확인되었다. 약동학적 임상시험에서, 이 약과 이트라코나졸(강력한 CYP3A4 저해제) 1일 2회 200mg를 병용투여 하였을 때 이 약의 노출(커브아래면적(AUC;Area Under the Curve)은 24%(90% CI 15, 35) 증가하였고 C<SUB>max</SUB>는 ‑20%(90% CI ‑27, ‑13) 감소)에는 임상적으로 유의한 영향을 미치지 않았다. 따라서 CYP3A4 저해제는 이 약의 노출량에 영향을 미치지 않는 것으로 보인다.
2) 이 약의 혈장 농도를 감소시킬 수 있는 활성 물질
강력한 CYP3A4 유도제는 오시머티닙의 노출을 감소시킬 수 있다. 환자를 대상으로 한 약동학적 임상시험에서, 리팜피신(21일간 600mg매일 투여)과 병용투여 시 이 약의 정상상태의 AUC는 ‑78%(90% CI ‑81, ‑76) 감소하였다. 이 약과 강력한 CYP3A 유도제(예, 페니토인, 리팜피신, 카바마제핀, 세인트존스워트(St. John’s Wort))와의 병용투여는 피하는 것이 권장된다.
3) 위산을 감소시키는 활성 성분이 이 약의 대사 미치는 영향
환자를 대상으로 한 약동학적 임상시험에서, 오메프라졸과의 병용투여는 이 약의 노출량에 대하여 임상적으로 관련 있는 변화를 야기하지 않았다. 위내 pH를 변화시키는 약물들은 제한 없이 이 약과 병용투여 할 수 있다.
4) 이 약에 의해 혈장 농도가 변할 수 있는 활성 성분
생체 외 시험에 근거하였을 때, 이 약은 BCRP 수송체의 경쟁적 저해제이다. 이 약은 breast cancer resistant protein(BCRP) 및 P‑gp 기질의 노출을 증가시킬 수 있다.
환자를 대상으로 한 약동학적 임상시험에서, 이 약과 로수바스타틴(민감한 BCRP 기질)의 병용투여는 로수바스타틴의 AUC와 C<SUB>max</SUB>를 각각 35%(90% CI 15, 57), 72%(90% CI 46, 103) 증가시켰다.
BCRP에 약물 분포가 의존적이며 치료영역이 좁은 약물을 병용 투여하는 환자는 초기용량 설정 시 약동학적 상호작용 가능성을 면밀히 고려하고 이 약 투여 중 병용약물의 노출량 증가에 기인하여 발생할 수 있는 내약성 변화의 징후에 대해 주의 깊게 모니터링 해야 한다.
환자를 대상으로 한 약동학적 임상시험에서, 이 약과 심바스타틴(민감한 CYP3A4 기질)의 병용투여는 심바스타틴의 AUC와 C<SUB>max</SUB>를 각각 ‑9% (90% CI ‑23, 8), ‑23 % (90% CI ‑37, ‑6) 감소시켰다. 이 변화는 작고, 임상적으로 유의하지 않은 것으로 보인다. CYP3A4 기질과의 임상적 약동학적 상호작용은 없는 것으로 보인다.
임상 약동학 시험에서, 이 약과와 펙소페나딘(PXR/P‑gp 기질)의 병용 투여는 펙소페나딘의 AUC 및 C<SUB>max</SUB>를 단회 투여 후 각각 56% (90% CI 35, 79) 및 76% (90% CI 49, 108) 증가시켰고 항정상태에서 각각 27% (90% CI 11, 46) 및 25% (90% CI 6, 48) 증가시켰다. P‑gp에 약물 분포가 의존적이며 치료 영역이 좁은 약물(예, 디곡신, 다비가트란, 알리스키렌)을 병용 투여하는 환자는 이 약 투여 중 병용 약물의 노출 증가에 기인한 내약성 변화의 징후에 대해 주의 깊게 모니터링 되어야 한다.
6. 임부 및 수유부에 대한 투여
1) 남성 및 여성에서의 피임
가임 여성은 이 약 투여 중 임신을 피하여야 한다. 환자는 이 약 투여 완료 후 여성은 최소 2개월, 남성은 최소 4개월 동안 효과적인 피임법을 계속 사용하도록 해야 한다.
2) 임부
이 약을 임부에게 투여한 자료는 없거나 제한적이다. 동물시험에서는 생식독성을 보였다.
작용기전 및 비임상시험 데이터에 근거할 때, 이 약은 임신한 여성에게 투여 시 태아 독성을 유발할 수 있다. 임신한 랫드에게 오시머티닙 투여 시, 사람에게 예상되는 노출량과 유사한 수준에서 배자 치사, 태자 성장 감소 및 신생자(neonate) 사망과 관련이 있는 것으로 나타났다. 이 약은 임부 및 피임을 하지 않고 임신할 가능성이 있는 여성에게 투여하지 않는다.
3) 수유부
오시머티닙 또는 그 대사체가 사람 모유로 분비되는지는 알려져 있지 않다.
임신한 랫드 및 초기 수유 중인 랫드에의 이 약의 투여는 성장율 감소와 신생자 사망(neonatal death)을 포함한 이상반응과 관련이 있었다. 이 약 또는 그 대사체가 동물의 모유로 분비되는지에 대한 정보는 충분하지 않다. 젖먹이 아이에게의 위험을 배제할 수 없다. 수유부에게 이 약 치료 중 수유를 중단하도록 해야 한다.
4) 수태능
사람의 수태능에 이 약이 미치는 영향에 대한 자료는 없다. 동물실험 결과 이 약이 남성 및 여성 생식기관에 영향을 주어 생식능력을 저하시킬 수 있는 것으로 나타났다.
7. 소아에 대한 투여
소아 환자에서 이 약의 안전성 ◦유효성은 확립되지 않았다.
8. 고령자에 대한 투여
집단 약동학 분석에서 연령은 오시머티닙의 노출량에 영향을 주지 않는 것으로 나타났다.
ADAURA, FLAURA, FLAURA2 및 AURA (이 약의 단독요법 군 (N = 1813))에서, 42 % 환자가 65세 이상이었고, 11%가 75세 이상이었다. 젊은 환자(65세 미만)와 비교하여, 65세 이상의 환자에서 시험약 용량 변경이 필요한 이상반응이 더 많이 보고되었다(일시중단 또는 용량감소)(14% vs 10%). 보고된 이상반응의 종류는 연령에 관계없이 유사하였다. 고령의 환자가 젊은 환자에 비해 3등급 이상의 이상반응을 더 많이 보고하였다(11% vs 9%). 고령의 환자와 젊은 환자 간 유효성에 전반적인 차이는 관찰되지 않았다.
9. 간장애 환자에 대한 투여
임상시험에 근거할 때 경증(Child Pugh A) 또는 중등증 간장애(Child Pugh B) 환자에서의 용량 조절은 필요하지 않다. 이와 유사하게, 집단 약동학 분석에 근거할 때, 경증의 간장애 환자(총빌리루빈≦정상 상한치[ULN] 이고 AST >ULN 또는 총빌리루빈이 1.0배의 ULN과 1.5배의 ULN 및 모든 수치의 AST 사이) 또는 중등증의 간장애 환자(총빌리루빈이 1.5배의 ULN과 3배의 ULN 및 모든 수치의 AST 사이)에서 용량 조절은 필요하지 않다.
중증의 간장애 환자에서 이 약의 안전성 ◦유효성은 확립되지 않았다.
10. 신장애 환자에 대한 투여
임상시험 및 집단 약동학 분석에 근거할 때, 경증, 중등증 또는 중증의 신장애 환자에게 용량조절은 필요하지 않다.
말기 신질환 환자 [Cockcroft and Gault 방정식에 의해 계산된 크레아티닌 청소율 (CLcr)이 15 mL/min 미만] 또는 투석 중인 환자에 대한 이 약의 안전성 ◦유효성은 확립되지 않았다. 중증 및 말기 신장애 환자 치료 시 주의해야 한다.
11. 과량투여시의 처치
이 약을 사용한 임상시험에서 제한된 수의 환자들이 이 약 1일 최대 240mg까지 용량 제한적 독성(dose limiting toxicities)없이 투여되었다. 이 연구들에서, 이 약을 1일 160mg 및 240mg 투여받은 환자들은 80mg을 투여받은 환자들보다 전형적인 EGFR‑유도 이상반응(주로 설사와 피부발진)의 빈도 및 중증도의 증가를 경험하였다.
이 약의 과량투여 시 특별한 치료방법이 없다. 의사는 일반적인 보조 조치를 실시하고 대증적으로 치료해야 한다.
12. 보관 및 취급상의 주의사항
1) 어린이의 손이 닿지 않는 곳에 보관한다.
2) 다른 용기에 바꾸어 넣는 것은 사고원인이 되거나 품질 유지 면에서 바람직하지 않으므로 이를 주의한다.
13. 전문가를 위한 정보
1) 약리작용
이 약은 티로신 키나제 억제제 (Tyrosine Kinase Inhibitor, TKI)이다. 이 약은 감작 돌연변이 (EGFRm) 및 TKI‑내성 돌연변이 T790M을 가진 표피 성장인자 수용체 (EGFRs)의 강력한 선택적 비가역적 경구 억제제이다.
In vitro 시험에서 이 약은 임상적으로 관련된 모든 EGFR 감작 돌연변이체 및 T790M 돌연변이체 비소세포폐암 (NSCLC) 다양한 세포주에 대하여 높은 효력 및 억제 활성을 나타내는 것을 입증하였다. (phospho‑EGFR에 대한 겉보기 IC<SUB>50</SUB>6 nM에서 54 nM) 이로 인해 세포 성장이 억제되는 반면, 야생형 세포주에서는 EGFR에 대한 활성이 유의하게 낮게 나타난다. (phospho‑EGFR에 대한 겉보기 IC<SUB>50</SUB>480 nM에서 1.9 μM) In vivo에서 이 약의 경구 투여는 EGFRm 및 T790M 모두의 NSCLC 이종이식 및 유전자 이식 마우스 폐 종양 모델에서 종양 축소를 나타난다.
심장 전기생리학
이 약의 QTc 간격 연장 가능성은 AURA2에서 오시머티닙 1일 80 mg을 투여 받은 210 명의 환자에서 평가되었다. QTc 간격에 대한 오시머티닙의 영향을 평가하기 위해 연속적 ECG를 단회 투여 및 정상상태에서 수집했다. 이 약의 약동학/약력학 분석은 80 mg에서 14 msec의 약물 관련 QTc 간격 연장을 상한 16 msec (90% CI)으로 예측했다.
2) 약동학적 정보
오시머티닙 약동학 파라미터는 건강한 시험대상자 및 NSCLC 환자들에서 확인되었다. 집단 약동학 분석에 근거하여, 오시머티닙 겉보기 혈장 청소율은 14.3 L/h이며, 겉보기 분포 용적은 918 L이고, 말단 반감기는 약 44시간이다. 이 약과 페메트렉시드 및 백금 기반 항암화학요법을 병용하여 치료한 환자의 약동학은 이 약 단독요법으로 치료받은 환자의 약물동태와 유사하다. AUC 및 Cmax 는 20에서 240mg 용량 범위에서 용량에 비례하여 증가하였다. 이 약을 1일 1회 투여한 결과, 약 3배의 축적을 나타내며, 정상 상태 노출은 투여 15일에 도달되었다. 정상 상태에서, 순환혈장농도는 일반적으로 24시간 투여 간격 동안 1.6배 범위 내에서 유지된다.
흡수
이 약을 경구 투여한 후, 오시머티닙의 최고 혈장 농도는 t<SUB>max</SUB>중앙값(최소‑최대)은 6시간 (3‑24)에 도달되었으며, 일부 환자들에서 처음 24시간 동안 여러 개의 피크가 관찰되었다. 이 약의 절대 생체 이용률은 70%이다. (90% CI 67, 73) 80 mg을 투여 받은 환자에 대한 임상 약동학 시험에 근거하여, 음식은 오시머티닙의 생체 이용률을 임상적으로 의미 있는 정도로 변경하지 않는다. (AUC 6% 증가 (90% CI ‑5, 19) 및 C<SUB>max</SUB>‑7%감소 (90% CI ‑19, 6)). 5일간 오메프라졸 투여에 의해 위장 pH가 상승한 건강한 시험대상자에서 80 mg 정제를 투여했을 때, 오시머티닙 노출은 영향을 받지 않았으며 (AUC 및 C<SUB>max</SUB>증가 각각 7% 및 2%), 노출비에 대한 90% CI는 80‑125% 한도 내에 포함되었다.
분포
모집단에서 평가된 오시머티닙의 정상 상태에서의 평균 분포 용적은 (V<SUB>ss</SUB>/F)918L이며, 이는 조직으로의 광범위한 분포를 시사한다. In vitro에서, 오시머티닙의 혈장 단백질 결합은 94.7%이다. (비결합 5.3%) 오시머티닙은 또한 랫드와 사람의 혈장 단백질, 사람 혈청 알부민 및 랫드와 사람 간세포에 공유 결합하는 것으로 확인되었다.
생체 내 변환
In vitro 시험에서 오시머티닙은 주로 CYP3A4 및 CYP3A5에 의해 대사되는 것을 보여준다. In vitro 시험에 근거하여, 두가지 약리학적 활성 대사체 (AZ7550 및 AZ5104)가 이 약을 경구 투여한 후 전임상 종의 혈장 및 사람에서 이후 확인되었다; AZ7550은 이 약과 유사한 약리학적 프로파일을 나타낸 반면, AZ5104는 돌연변이 및 야생형 EGFR 모두에서 더 큰 효력을 보였다. 환자들에게 이 약을 투여한 후 두 대사체 모두 혈장에서 서서히 나타났으며, t<SUB>max</SUB>중앙값 (최소‑최대)은 각각 24 (4‑72) 및 24 (6‑72) 시간이었다. 사람 혈장에서, 모성분 오시머티닙은 0.8%를 차지하였고, 두 대사체는 총 방사능의 0.08% 및 0.07%에 기여하였으며, 방사능의 대부분은 혈장 단백질에 공유 결합하였다. AUC에 근거한 AZ5104 및 AZ7550의 기하 평균 노출은 정상 상태에서 오시머티닙 노출의 각각 약 10%였다.
오시머티닙의 주요 대사 경로는 산화 및 탈알킬화였다. 사람의 뇨 및 분변 통합 검체에서 최소 12개의 성분들이 관찰되었으며, 5개 성분은 투여량의 >1%를 차지하였고, 이 중 미변화 오시머티닙, AZ5104, 및 AZ7550은 투여량의 약 1.9, 6.6, 및 2.7%를 차지한 반면, cysteinyl 부가생성물 (M21), 및 알려지지 않은 대사체 (M25)는 투여량의 각각 1.5% 및 1.9%를 차지하였다.
In vitro 시험에 근거하여 오시머티닙은 임상적으로 관련된 농도에서 CYP 3A4/5의 경쟁적 억제제이지만, CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 및 2E1 에 대해서는 아니다. In vitro 시험에 근거하여 오시머티닙은 간에서 임상적으로 관련된 농도에서 UGT1A1 및 UGT2B7의 억제제가 아니다. UGT1A1의 장에서의 억제는 가능하지만, 임상적 영향은 알려지지 않았다.
배설
20 mg 단회 경구 투여 후, 투여량의 67.8%가 분변으로 회수된 반면 (모성분으로 1.2%), 투여량의 14.2% (모성분으로 0.8%)는 84일의 표본 수집 시까지 소변에서 발견되었다. 미변화된 오시머티닙은 배설의 약 2%를 차지하였으며, 0.8%는 소변으로, 1.2%는 분변으로 배설되었다.
특수 집단
집단 기반 PK 분석에서 (n=1367), 예측된 정상 상태 노출 (AUC<SUB>ss</SUB>)과 환자의 연령 (범위: 25‑91세), 성별 (65% 여성), 인종 (백인, 아시아인, 일본인, 중국인 및 비아시아‑비백인 환자), 치료 차수 및 흡연 상태 (n=34 현재 흡연자, n=419 과거 흡연자) 간에 임상적으로 유의한 관련성은 확인되지 않았다. 집단 PK 분석에서 체중이 유의한 공변량으로 나타났으며, 체중 중앙값 61 kg의 AUC<SUB>ss</SUB>와 비교했을 때, 88 kg에서 43 kg 체중 범위에서 오시머티닙 AUC<SUB>ss</SUB>변화가 20% 미만으로 예상된다. (95%‑5% 사분위) 체중의 극단을 고려하여 43 kg 미만에서 88 kg 초과까지 AZ5104 대사체 비는 11.8% 에서 9.6% 범위이며, AZ7550의 경우 그 범위는 12.8%에서 8.1%이다. 집단 PK 분석에 근거하여 혈청 알부민은 유의한 공변량으로 확인되었으며, 베이스라인 알부민 중앙값 39 g/L에 대한 AUC<SUB>ss</SUB>와 비교했을 때 알부민 범위 29에서 46 g/L의 알부민 범위에서 오시머티닙 AUCss 변화는 30% 미만으로 예상된다. (95% ‑ 5% 사분위수) 체중 또는 베이스라인 알부민 차이에 의한 이러한 노출 변화는 임상적으로 관련이 없는 것으로 고려된다.
뇌 전이 환자
뇌 전이가 있는 EGFR 돌연변이 양성 NSCLC 환자 (n=4)를 대상으로 한 마이크로도즈 PET 연구에서 오시머티닙의 뇌 침투 및 분포(중앙값 T<SUB>max</SUB>: 22분;평균C<SUB>max</SUB>: 투여된 용량의 1.5%가 뇌에 도달)는 건강한 지원자 연구에서 관찰된 것과 유사했다 (n=7; 중앙값 T<SUB>max</SUB>: 11분; 평균 C<SUB>max</SUB>: 투여된 용량의 2.2%가 뇌에 도달).
3) 임상적 유효성 및 안전성
이전 보조 화학 요법을 받았거나 받지 않은 EGFR 변이 양성 비소세포폐암의 보조 치료 ‑ ADAURA
이전 보조 화학 요법을 받았거나 받지 않은 완전 종양 절제술을 받은 EGFR 변이 양성 (엑손 19 결손 또는 L858R 치환 변이) 비소세포폐암 환자의 보조 치료에 대한 이 약의 유효성 및 안전성은 무작위 이중 맹검 위약 대조 시험 (ADAURA)에서 입증되었다.
절제 가능한 종양 (IA기 제외)이 있는 환자들은 중앙 실험실에서 생검 또는 수술 검체를 사용하여 전향적으로 실시된 cobas EGFR Mutation Test에 의해 확인된 EGFR 엑손 19 결손 또는 엑손 21 L858R 치환 변이가 있어야 했다.
환자들은 무작위 배정되어 (1:1) 이 약 80 mg 경구 1일 1 회 또는 위약을 수술에서 회복된 후 투여 받고 제공된 경우 표준 보조 화학 요법을 받았다. 보조 화학 요법을 받지 않은 환자들은 수술 후 10 주 이내에, 보조 화학 요법을 받은 환자들은 수술 후 26 주 이내에 무작위 배정되었다. 무작위 배정은 돌연변이 유형 (엑손 19 결손 또는 엑손 21 L858R 치환 변이), 민족성 (아시아인 또는 비아시아인) 및 AJCC 7판에 따른 pTNM (IB 또는 II 또는 IIIA) 기반 병기 별로 계층화되었다. 질병이 재발하거나 수용할 수 없는 독성이 나타날 때까지 또는 3년 동안 투여를 받았다.
주요 유효성 결과 측정은 연구자 평가에 따른 무병 생존 기간 (disease‑free survival, DFS)이었다. 추가적 유효성 결과 측정은 DFS 비율, 전체 생존기간 (overall survival, OS), OS 비율 및 건강 관련 삶의 질 (HRQL) SF‑36이 악화되는 시간을 포함한다.
총 682 명의 환자가 이 약 (n = 339) 또는 위약 (n = 343)에 무작위 배정되었다. 중앙 연령은 63세 (30‑86세 범위)였고 11%는 75세 이상; 70%는 여성, 64%는 아시아인, 72%는 흡연 미경험자였다. 베이스라인 WHO 수행 능력은 0 (64%) 또는 1 (36%)이었다; 31%는 IB기, 34% II기, 그리고 35%는 IIIA기였다. EGFR 돌연변이 상태와 관련하여, 55%는 엑손 19 결손이었고 45%는 엑손 21 L858R 치환 변이였다; 9명의 환자 (1%)는 de novo T790M 변이도 있었다. 환자의 대다수 (60%)는 무작위 배정 전 보조 화학 요법을 받았다 (26% IB; 71% IIA; 73% IIB; 80% IIIA).
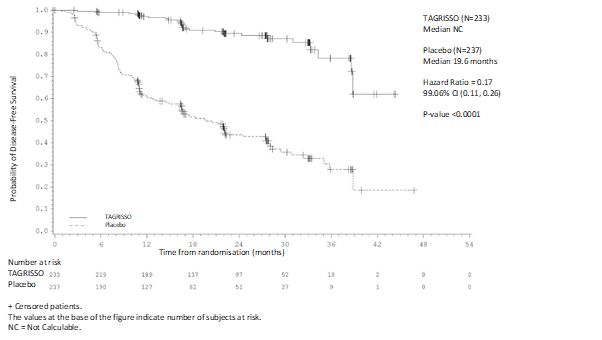
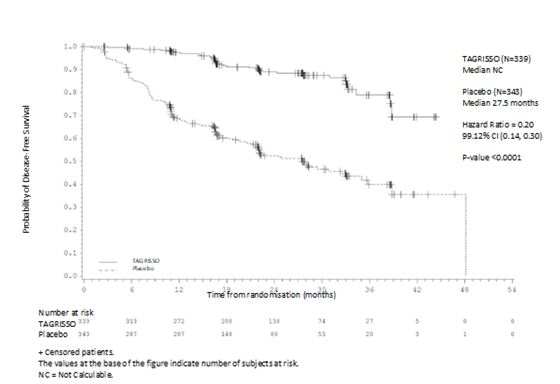
II‑IIIA기 집단과 전체 집단 (IB‑IIIA)에 대해 DFS 분석을 했다. ADAURA는 위약으로 치료받은 환자 대비 이 약으로 치료받은 환자에 대해 질병 재발 또는 사망의 위험이 통계적으로 유의하고 임상적으로 의미있는 감소함을 입증하였다. 이 약으로 치료받은 II‑IIIA기 질환 환자들은 위약 대비 질병 재발 또는 사망의 위험이 83% 감소했다 (중앙값은 각각 계산되지 않음 (not calculated, NC) 및 19.6개월, HR=0.17, 99.06% CI:0.11, 0.26; P<0.0001). 위약 대비 이 약으로 치료 받은 전체 집단 (IB‑IIIA)은 질병 재발 또는 사망의 위험이 80% 감소한 것으로 나타났다 (중앙값은 각각 NC 및 27.5개월, HR=0.20, 99.12% CI:0.14, 0.30; P<0.0001).
이 약에서 질병이 재발한 환자는 37명이었다. 가장 흔하게 보고된 재발 부위는 다음과 같다; 폐 (19 명); 림프절 (10명) 및 CNS (5명). 위약으로 질병이 재발한 환자는 157명이었다. 가장 흔하게 보고된 부위는 다음과 같다; 폐 (61명); 림프절 (48명) 및 CNS (34명).
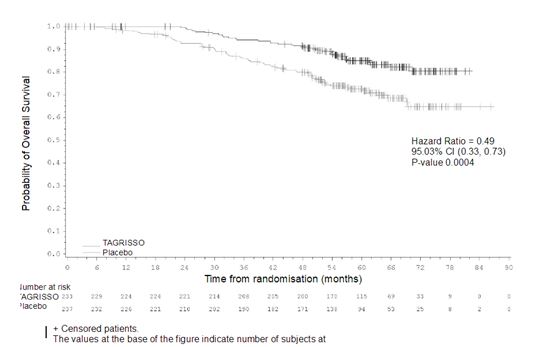
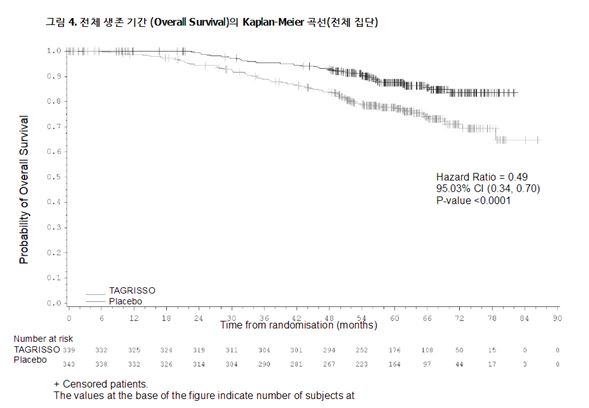
OS의 최종 분석은 II‑IIIA기 집단 (21.3% 성숙; HR=0.49; 95.03% CI: 0.33, 0.73; p‑값=0.0004) 및 전체 집단 (IB‑IIIA; 18.2% 성숙; HR=0.49; 95.03% CI: 0.34, 0.70; p 값 < 0.0001) 둘 모두에 대해 위약과 비교하여 이 약을 투여받은 환자의 51% 사망 위험 감소를 보이며 임상적으로 의미 있고 통계적으로 크게 유의한 OS 개선을 입증하였다. 전체 집단 (IB‑IIIA)에서, 중도절단된 환자의 추적관찰 시간의 중앙값은 두 치료군 모두 61.5개월이었다.
연구자 평가에 따른 ADAURA의 유효성 결과는 표 8 및 9에 요약되어 있으며, II‑IIIA기 환자 및 전체 집단 (IB‑IIIA)의 DFS 및 OS 에 대한 Kaplan‑Meier 곡선은 그림 1부터 그림 4에 제시하였다.
표 8. 연구자 평가에 따른 II‑IIIA기 환자에서의 유효성 결과
|
유효성 지표 |
이 약 (N=233) |
위약 (N=237) |
|
무병 생존 기간 (Disease Free Survival) | ||
|
사건 수 (%) |
26 (11.2) |
130 (54.9) |
|
질병 재발 (%) |
26 (11.2) |
129 (54.4) |
|
사망 (%) |
0 |
1 (0.4) |
|
DFS 중앙값, 개월(95% CI) |
NC (38.8, NC) |
19.6 (16.6, 24.5) |
|
HR (99.06% CI); P-값a |
0.17 (0.11, 0.26); <0.0001 | |
|
12개월 시점 DFS 비율 (%) (95% CI) |
97.2 (93.9, 98.7) |
60.8 (54.1, 66.8) |
|
24개월 시점 DFS 비율 (%) (95% CI) |
89.5 (84, 93.2) |
43.6 (36.5, 50.6) |
|
36개월 시점 DFS 비율 (%) (95% CI)b |
78.3 (64.5, 87.3) |
27.9 (18.9, 37.6) |
|
전체 생존 기간 (Overall Survival) | ||
|
사망 수 (21% 성숙) |
35 (15.0) |
65 (27.4) |
|
OS 중앙값, 개월 (95% CI) |
NC (NC, NC) |
NC (NC, NC) |
|
HR (95.03% CI); P-값c |
0.49 (0.33, 0.73); 0.0004 | |
|
24개월 시점 OS 비율 (%) (95% CI)d |
99.5 (96.8, 99.9) |
92.6 (88.3, 95.3) |
|
36개월 시점 OS 비율 (%) (95% CI)d |
94.1 (90.1, 96.5) |
85.9 (80.6, 89.8) |
|
48개월 시점 OS 비율 (%) (95% CI)d |
91.0 (86.3, 94.1) |
79.9 (74.0, 84.6) |
|
60개월 시점 OS 비율 (%) (95% CI)d |
85.0 (79.3, 89.2) |
72.6 (66.0, 78.1) |
DFS 결과는 연구자 평가에 근거한 것이다.
HR <1은 이 약에 유리하다.
DFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 22.1개월이었고 위약을 투여 받은 환자에서 14.9개월이었다.
중도절단된 환자의 OS에 대한 추적관찰 시간의 중앙값은 이 약 투여 군(stage II‑IIIA 집단)에서 61.7개월, 위약 군에서 60.4개월이었다.
DFS 결과는 1차 분석(2020년 1월 17일)에서 나왔다. OS 결과는 최종 분석(2023년 1월 27일)에서 나왔다.
a 중간분석 (33% 성숙)을 위해 조정된 p‑값 <0.0094이 통계적 유의성을 달성하기 위해 필요했다.
b 36개월 시점에 추적 관찰 환자 (patient at risk) 수는 이 약 투여군은 18명이고, 위약 군은 9명이었다.
c 중간 분석을 위해 조정된 통계적 유의성을 달성하려면 p‑값 <0.0497이 필요했다.
d Kaplan‑Meier 방법으로 계산했다.
그림 1. 연구자 평가에 따른 무병 생존 기간 (II‑IIIA기 환자)의 Kaplan‑Meier 곡선
|
유효성 지표 |
이 약 (N=339) |
위약 (N=343) |
|
무병 생존 기간 (Disease Free Survival) | ||
|
사건 수 (%) |
37 (10.9) |
159 (46.4) |
|
질병 재발 (%) |
37 (10.9) |
157 (45.8) |
|
사망 (%) |
0 |
2 (0.6) |
|
DFS 중앙값, 개월 (95% CI) |
NC (NC, NC) |
27.5 (22.0, 35.0) |
|
HR (99.12% CI); P -값a |
0.20 (0.14, 0.30); <0.0001 | |
|
12개월 시점 DFS 비율 (%) (95% CI) |
97.4 (94.9, 98.7) |
68.5 (63.2, 73.2) |
|
24개월 시점 DFS 비율 (%) (95% CI) |
89.1 (84.5, 92.4) |
52.4 (46.4, 58.1) |
|
36개월 시점 DFS 비율 (%) (95% CI)b |
78.9 (68.7, 86.1) |
40.0 (32.1, 47.8) |
|
전체 생존 기간 (Overall Survival) | ||
|
사망 수 (18% 성숙) |
42 (12.4) |
82 (23.9) |
|
OS 중앙값, 개월 (95% CI) |
NC (NC, NC) |
NC (NC, NC) |
|
HR (95.03% CI); P-값c |
0.49 (0.34, 0.70); <0.0001 | |
|
24개월 시점 OS 비율 (%) (95% CI)d |
99.4 (97.5, 99.8) |
94.3 (91.2, 96.3) |
|
36개월 시점 OS 비율 (%) (95% CI)d |
95.3 (92.3, 97.1) |
88.8 (84.9, 91.8) |
|
48개월 시점 OS 비율 (%) (95% CI)d |
92.8 (89.4, 95.2) |
83.9 (79.4, 87.4) |
|
60개월 시점 OS 비율 (%) (95% CI)d |
87.6 (83.3, 90.9) |
77.7 (72.7, 81.9) |
HR (Hazard Ratio)=위험비; CI (Confidence Interval)=신뢰 구간; NC (Not Calculable)=계산 불가능
DFS 결과는 연구자 평가에 근거한 것이다.
HR <1은 이 약에 유리하다.
DFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 22.1개월이었고 위약을 투여 받은 환자에서 16.6개월이었다.
중도절단된 환자의 OS에 대한 추적관찰 시간의 중앙값은 이 약 투여 군 및 위약 군에서 모두 61.5개월이었다.
DFS 결과는 1차 분석(2020년 1월 17일)에서 나왔다. OS 결과는 최종 분석(2023년 1월 27일)에서 나왔다.
a 중간분석 (29% 성숙)을 위해 조정된 p‑값 < 0.0088이 통계적 유의성을 달성하기 위해 필요했다.
b 36개월 시점에 추적 관찰 환자 (patient at risk) 수는 이 약 투여 군은 27명이고, 위약 군은 20명이었다.
c 중간 분석을 위해 조정된 통계적 유의성을 달성하려면 p‑값 < 0.0497이 필요했다.
d Kaplan‑Meier 방법으로 계산했다.
그림 3. 연구자 평가에 따른 무병 생존 기간 (전체 집단)의 Kaplan‑Meier 곡선
위약 환자 대비 이 약 환자에 대한 CNS DFS (CNS 재발 또는 사망까지의 시간)의 탐색적 분석에서 임상적으로 의미있는 개선이 전체 집단에서 HR 0.18 (95% CI:0.10, 0.33; P <0.0001)로 관찰되었으며, 이는 위약 대비 이 약 군에서 CNS 질환 재발 또는 사망의 위험이 82% 감소함을 나타낸다.
환자 보고 결과
ADAURA에서 건강 관련 삶의 질 (HRQL)은 Short Form (36) Health Survey version 2 (SF‑36v2) 설문지를 사용하여 평가되었다. SF‑36v2는 치료가 완료되거나 중단될 시까지 무작위 배정과 관련하여 12주, 24주, 그 후 24주마다 관리되었다. 전반적으로, HRQL은 두 군 모두에서 유지되었으며, II‑IIIA기 집단에서 75%를 넘는 환자가 SF‑36의 물리적 구성요소에서 임상적으로 의미있는 악화 또는 사망을 경험하지 않았거나 (이 약 대 위약에 대해 75.1% 대 83.5%) 또는 SF‑36의 정신적 구성요소에서 임상적으로 의미있는 악화 또는 사망 (이 약 대 위약에 대해 77.7% 대 78.1%)을 경험하지 않았다. 이 약 군에서 SF‑36의 물리적 구성요소 또는 사망에 대한 약화 시간 (time to deterioration, TTD)이 짧은 경향이 관찰되었으며 (HR=1.43, 95% CI:0.96, 2.13), 두 군 모두에서 중앙값 TTD에 도달하지 않았다. SF‑36의 정신적 구성요소 또는 사망에 대한 TTD에서 군 간의 차이는 없었으며 (HR=0.90, 95% CI: 0.61, 1.33), 중앙값 TTD는 이 약 군에서 39.0개월 (95% CI: NC, NC)이고 위약 군에서는 도달하지 않았다.
절제 불가능한 국소 진행성(III기) EGFR 돌연변이 양성 비소세포폐암 – LAURA
확정적 백금‑기반 화학 방사선 요법 중 또는 후 진행되지 않은, EGFR 돌연변이 양성의 절제 불가능한 국소 진행성(III기) 비소세포폐암 환자의 치료에 대한 이 약의 유효성 및 안전성은 무작위 배정, 이중 눈가림, 위약 대조 시험(LAURA)에서 입증되었다. 환자들은 동시적 화학 방사선 치료(CCRT)나 순차적 화학 방사선 치료(SCRT) 요법을 받았으며, 여기에서 최소 2주기 또는 주 5 회 용량의 백금‑기반 화학 요법과 총 60 Gy ±10% (54 Gy ‑ 66 Gy) 용량의 방사선 치료가 무작위 배정 전 6주 이내에 완료되었다. 환자 종양 조직 검체는 공인/승인된 시험을 이용하여 중앙 또는 현지 검사로 확인된 EGFR 엑손 19 결손 또는 엑손 21 L858R 변이가 있어야 했다. 확정적 백금‑기반 화학 방사선 요법 중 또는 후 2등급 이상의 폐염증이 있거나 또는 화학 방사선 요법 이전에 간질성 폐염증의 병력이 있는 환자는 제외되었다.
환자들은 이 약 80 mg을 1일 1회 경구로(n=143) 또는 위약(n=73)을 투여받도록 무작위 배정되었다(2:1). 무작위 배정은 선행 화학 방사선 치료 전략(CCRT vs SCRT), 화학 방사선 치료 전 종양 병기 결정(IIIA vs IIIB/IIIC), 및 중국 코호트에 따라 층화되었다. 환자들은 치료에 대한 불내성 또는 확인된 질병 진행까지 시험 치료를 계속 받아야 했다. 이 후, 모든 환자는 치료 의사의 소견에 따라 기대되는 임상적 유익성이 있는 경우 현지 임상진료에 따라 라벨이 공개된 이 약을 제공받았다.
일차 유효성 평가변수는 눈가림된 독립적 중앙 검토(BICR)로 평가된 PFS였다. 추가 유효성 평가변수에는 CNS PFS, OS, ORR, DoR, 사망 또는 원격 전이까지 걸린 시간(TTDM), 첫 번째 후속 치료 시작 후 두 번째 PFS (PFS2), 첫 번째 후속 치료 또는 사망까지 걸린 시간(TFST), 및 두 번째 후속 치료 또는 사망까지 걸린 시간(TSST)이 포함되었다. CNS PFS, ORR, DoR, 및 TTDM은 모두 BICR로 평가되었다. 또한 환자 보고 결과도 평가되었다.
전체 시험대상군의 베이스라인 인구학적 및 질병 특성은 다음과 같았다: 연령 중앙값 63세(범위 36‑84세), ≧75세(13%), 여성(61%), 아시아인(82%), 백인(14%), 흡연력이 없는 사람(70%). 베이스라인 WHO 수행 능력 상태는 0 (51%) 또는 1 (49%)이었다; 환자의 35%는 IIIA기, 49%는 IIIB기, 그리고 16%는 IIIC기 비소세포폐암이었다. 환자의 조직학적 특성으로는 96%는 비편평세포암이었고, 4%는 편평세포암이었다. EGFR 돌연변이 상태와 관련해서는 54%가 엑손 19 결손이었고 45%가 엑손 21 L858R 변이였다. 무작위 배정 전에 환자의 89%는 CCRT를 받았고 11%는 SCRT를 받았다. 모든 환자는 백금‑기반 화학 요법(55%는 카보플라틴‑기반 화학 요법, 44%는 시스플라틴‑기반 화학 요법)을 받았다. 총 방사선량의 중앙값은 두 군에서 모두 환자에서 60 Gy였다.
백금‑기반 화학 방사선 요법 후 이 약의 치료는 위약 대비 PFS의 통계적으로 유의하고 임상적으로 유의미한 개선을 가져왔다(56% 성숙; HR=0.16; 95% CI: 0.10, 0.24; P<0.001, 각각 중앙값 39.1개월 및 5.6개월). 6, 12, 18, 24 및 36개월에서 이 약으로 치료 받은 환자의 무진행 생존 비율(각각 84%, 74%, 67%, 65% 및 58%)은 위약으로 치료받은 환자(각각 45%, 22%, 14%, 13% 및 10%)의 무진행 생존 비율보다 더 컸다.
임상시험 계획서에 따라, 모든 환자는 베이스라인 자기 공명 영상(MRI) 뇌 스캔을 받았고, 치료 중에 1명을 제외하고 모두 예정된 MRI 뇌 스캔을 받았다. 위약과 비교해 이 약으로 치료받은 환자에서 CNS PFS (신경방사선학자 BICR 평가에 근거)의 명목상 통계적으로 유의하고 임상적으로 유의미한 개선이 있었다(27% 성숙; HR=0.17; 95% CI: 0.09, 0.32; P<0.001 [명목]). 위약군에 비해 이 약 군에서 더 낮은 비율의 환자가 신경방사선학자 검토에 따라 새로운 CNS 병변이 있었다(각각 17/143 [12%] vs 26/73 [36%]).
전체 생존 기간의 중간 분석에서, 이 약에 유리한 긍정적 경향이 있었지만(20% 성숙; HR=0.81; 95% CI: 0.42, 1.56; P=0.530) 통계적으로 유의하지 않았다.
LAURA 임상시험에서 얻은 유효성 결과는 표 10에 요약되어 있으며, PFS와 CNS PFS에 대한 Kaplan‑Meier 곡선은 각각 그림 5 및 6에 나타내었다.
표 10. LAURA에서 얻은 유효성 결과
|
유효성 지표 |
이 약 (N=143) |
위약 (N=73) |
|
무진행 생존 기간 (Progression-Free Survival) | ||
|
사건 수(%) |
57 (40) |
63 (86) |
|
PFS 중앙값, 개월(95% CI) |
39.1 (31.5, NC) |
5.6 (3.7, 7.4) |
|
HR (95% CI); P-값 |
0.16 (0.10, 0.24); P<0.001 | |
|
CNS 무진행 생존 기간 (CNS Progression-Free Survival) | ||
|
사건 수(%) |
29 (20) |
30 (41) |
|
CNS PFS 중앙값, 개월(95% CI) |
NC (NC, NC) |
14.9 (7.4, NC) |
|
HR (95% CI); P-값 |
0.17 (0.09, 0.32); P<0.001 | |
|
12개월차 CNS 무진행 생존율, % (95% CI) |
87 (79, 92) |
53 (38, 66) |
|
24개월차 CNS 무진행 생존율, % (95% CI) |
83 (75, 89) |
43 (28, 58) |
|
전체 생존 기간 (Overall survival) | ||
|
사망 수(%) |
28 (20) |
15 (21) |
|
OS 중앙값, 개월(95% CI) |
54.0 (46.5, NC) |
NC (42.1, NC) |
|
HR (95% CI); P-값 |
0.81 (0.42, 1.56); P=0.530a | |
|
객관적 반응률b | ||
|
반응 수(n), 반응률 % (95% CI) |
82 57 (49, 66) |
24 33 (22, 45) |
|
오즈비(95% CI); P-값c |
2.77 (1.54, 5.08); P<0.001 | |
|
반응 기간(Duration of Response, DoR)b | ||
|
DoR 중앙값, 개월(95% CI)d |
36.9 (30.1, NC) |
6.5 (3.6, 8.3) |
|
사망 또는 원격 전이까지 걸린 시간(TTDM) | ||
|
환자 수(%) |
33 (23) |
31 (43) |
|
TTDM 중앙값, 개월(95% CI) |
NC (39.3, NC) |
13.0 (9.0, NC) |
|
HR (95% CI); P-값 |
0.21 (0.11, 0.38); P<0.001 | |
|
첫 번째 후속 치료 시작 후 두 번째 PFS (PFS2) | ||
|
PFS2 사례 수(%) |
34 (24) |
24 (33) |
|
PFS2 중앙값, 개월(95% CI) |
48.2 (44.4, NC) |
47.4 (28.2, NC) |
|
HR (95% CI); P-값 |
0.62 (0.35, 1.08); P=0.088 | |
|
무작위 배정부터 첫 번째 후속 치료 또는 사망까지의 시간(TFST) | ||
|
첫 번째 후속 치료를 받거나 사망한 환자 수(%) |
53 (37) |
61 (84) |
|
TFST 중앙값, 개월(95% CI) |
43.8 (38.9, NC) |
9.5 (6.6, 11.5) |
|
HR (95% CI); P-값 |
0.13 (0.08, 0.21); P<0.001 | |
|
무작위 배정부터 두 번째 후속 치료 또는 사망까지의 시간(TSST) | ||
|
두 번째 후속 치료를 받거나 사망한 환자 수(%) |
32 (22) |
24 (33) |
|
TSST 중앙값, 개월(95% CI) |
NC (44.4, NC) |
47.4 (34.1, NC) |
|
HR (95% CI); P-값 |
0.51 (0.28, 0.91); P=0.022 | |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, CNS PFS, ORR, DoR, 및 TTDM 결과는 BICR에 따라 평가하였다.
모든 환자에서 PFS에 대한 추적관찰 시간 중앙값은 이 약 군에서 22.0개월, 위약군에서 5.6개월이었다.
HR <1 및 오즈비 >1은 이 약에 유리하다.
a 중간 분석(20% 성숙)에 대해 보정한 결과 통계적 유의성에 도달하기 위해서는 p 값<0.00036이 필요했다.
b 확인되지 않은 응답을 기반으로 했다.
c 분석은 화학 요법 전 병기(IIIA vs IIIB/IIIC)에 따라 계층화된 로지스틱 회귀 분석을 사용하여 수행되었다.
d Kaplan‑Meier 방법으로 계산했다.
그림 5. LAURA에서 BICR로 평가된 PFS에 대한 Kaplan‑Meier 곡선
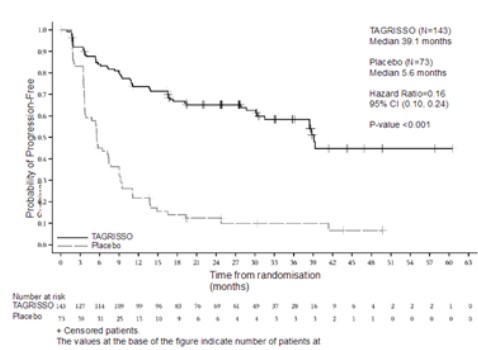
위약 대비 이 약의 PFS 유익성은 성별, 무작위 배정 시점의 연령, 흡연력, 민족성, 선행 화학 방사선 치료 전략(CCRT vs SCRT), 화학 방사선 치료 전 병기(IIIA vs IIIB/IIIC), 선행 화학 방사선 치료에 대한 반응, 및 EGFR 돌연변이 유형(엑손 19 결손 또는 L858R)을 포함하여 분석된 사전 정의된 모든 하위군에서 일관적이었다.
그림 6. LAURA에서 BICR로 평가된 CNS PFS의 Kaplan‑Meier 곡선
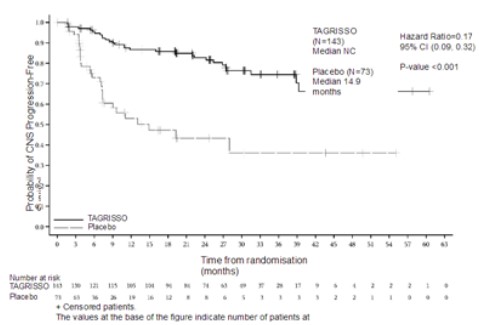
이 약에 무작위 배정된 환자는 위약에 무작위 배정된 환자에 비해 PFS2, TFST 및 TSST에서 임상적으로 유의미한 개선이 있었다. 이러한 진행 후 평가변수의 분석은 이 약으로 교차 투여 수준이 높음에도 불구하고 PFS 유익성이 후속 차수 치료를 통해 유지됨을 입증하였다.
환자 보고 결과
환자 보고 증상 및 HRQL 자료는 EORTC QLQ‑C30 (C30) 및 폐암 모듈 EORTC QLQ‑LC13 (LC13)을 이용하여 전자적으로 수집되었다. 베이스라인에서 환자 보고 증상, 신체적 기능 및 전반적인 건강 상태/삶의 질(GHS/QoL)은 이 약 군과 위약군 간에 유사하였다. 환자가 보고한 5가지 주요 폐암 및 치료 관련 증상(기침, 호흡 곤란, 흉통, 피로, 및 식욕 상실)과 신체적 기능 영역 및 GHS/QoL의 베이스라인 대비 전체 평균 변화에서 임상적으로 유의미한 차이는 없었다. 치료군 간에 이러한 환자 보고 증상, 신체적 기능 또는 GHS/QoL의 악화 위험에서 유의미한 차이는 관찰되지 않았다.
이전에 치료 받은 적 없는 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 – FLAURA –단독요법
진행성 질환에 대하여 이전에 전신 치료를 받지 않은 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 치료 시 이 약의 유효성 및 안전성이 무작위배정, 이중 눈가림, 활성 대조 시험(FLAURA)에서 입증되었다.
환자들은 이 약 (n=279, 80 mg 1일 1회 경구 투여) 또는 EGFR TKI 대조약(n=277; 게피티닙 250 mg 1일 1회 경구 투여 또는 엘로티닙 150 mg 1일 1회 경구 투여)을 투여 받도록 1:1로 무작위 배정 되었다. 무작위 배정은 EGFR 변이 유형(Ex19del 또는 L858R) 및 민족성(아시아인 또는 비아시아인)에 따라 층화되었다. 환자들은 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더 이상 임상적 유익성을 경험하지 않을 때까지 시험 치료를 투여 받았다. EGFR TKI 대조약을 투여 받은 환자의 경우, 진행이 발생한 후 종양 샘플 검사 시 T790M 변이에 양성인 경우 이 약 공개라벨으로 교차가 허용되었다.
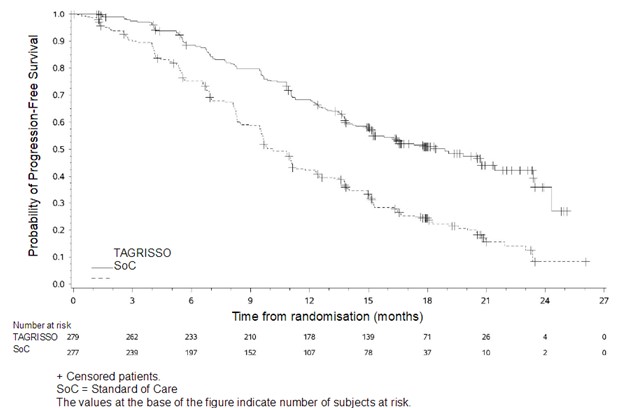
이 약은 EGFR TKI 대조약과 비교하여 임상적으로 의미 있고 통계적으로 크게 유의한 PFS 개선을 입증하였다 (중앙값 각각 18.9 개월 및 10.2 개월, HR=0.46, 95% CI: 0.37, 0.57; P<0.0001). 연구자 평가에 의한 FLAURA에서의 유효성 결과가 표 11에 요약되어 있으며, PFS에 대한 Kaplan‑Meier 곡선을 그림 7에 나타내었다. 전체 생존 기간 최종 분석 (58% 성숙)은 EGFR TKI 대조약과 비교하여 이 약에 무작위 배정된 환자에서 0.799의 HR (95% CI: 0.641, 0.996; P = 0.0462)로 통계적으로 유의한 개선 및 임상적으로 의미 있는 보다 긴 중앙 생존 기간을 입증했다 (표 11 및 그림 8). 12개월, 18개월, 24개월 및 36개월 시점에 생존한 환자 비율은 이 약 (각각 89%, 81%, 74% 및 54%) 치료 시 EGFR TKI 대조약 (각각 83%, 71%, 59% 및 44%) 치료에 비해 높았다.
표 11. 연구자 평가에 따른 FLAURA에서의 유효성 결과
|
유효성 지표 |
이 약 (N=279) |
EGFR TKI 대조약 (게피티닙 또는 엘로티닙) (N=277) |
|
무진행 생존 기간(Progression-Free Survival) | ||
|
사건 수 (62% 성숙) |
136 (49) |
206 (74) |
|
PFS 중앙값, 개월 (95% CI) |
18.9 (15.2, 21.4) |
10.2 (9.6, 11.1) |
|
HR (95% CI); P-값 |
0.46 (0.37, 0.57); P<0.0001 | |
|
전체 생존 기간(Overall Survival) | ||
|
사망 수, (58% 성숙) |
155 (56) |
166 (60) |
|
OS 중앙값, 개월 (95% CI) |
38.6 (34.5, 41.8) |
31.8 (26.6, 36.0) |
|
HR (95% CI); P-값 |
0.799 (0.641, 0.997); P=0.0462a | |
|
객관적 반응률(Objective Response Rate)*b | ||
|
반응자 수 (n), 반응률 % (95% CI) |
223 80 (75, 85) |
210 76 (70, 81) |
|
오즈비 (95% CI); P-값 |
1.3 (0.9, 1.9); P=0.2421 | |
|
반응 기간 (Duration of Response, DoR)* | ||
|
DoR 중앙값, 개월 (95% CI) |
17.2 (13.8, 22.0) |
8.5 (7.3, 9.8) |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, ORR, DoR 결과는 RECIST 연구자 평가에 근거한 것이다.
*확인되지 않은 반응에 근거한 것이다.
PFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 15.0개월이었고 EGFR TKI 대조약을 투여 받은 환자에서 9.7개월이었다.
OS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 35.8개월이었고 EGFR TKI 대조약을 투여 받은 환자에서 27.0개월이었다.
PFS, ORR, DoR 결과는 자료마감일 (data cut‑off) 2017년 6월 12일이다. OS 결과는 자료마감일 2019년 6월 25일이다.
HR <1 및 오즈비 >1은 이 약에 유리하다.
a 중간 분석(25% 성숙)을 위해 조정된 통계적 유의성을 달성하려면 p‑값 <0.0495이 필요했다
b 눈가림된 독립적 중앙 검토(BICR)에서 평가한 ORR 결과는 연구자 평가에서 보고된 것과 일치하였다; BICR 평가에 따른 ORR은 이 약에서 78% (95% CI: 73, 83)였고 EGFR TKI 대조약에서 70% (95% CI: 65, 76)였다.
그림 7. FLAURA에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선

FLAURA2 – 병용요법
진행성 질환에 대하여 이전에 전신 치료를 받지 않은 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 치료 시 페메트렉시드와 백금 기반 항암화학요법과 병용하는 이 약의 유효성 및 안전성이 무작위배정, 공개, 활성 대조 시험 (FLAURA2)에서 입증되었다. 환자의 종양 조직 샘플은 현지 또는 중앙 검사로 확인된, EGFR TKI 민감도와 관련이 있는 것으로 알려진 두 가지 일반적인 EGFR 변이 (Ex19del 또는 L858R) 중 하나가 있어야 했다.
환자들은 다음의 치료군 중 하나에 무작위 배정되었다 (1:1):
‧ 이 약 (80 mg) 1일 1회 경구 투여 시, 페메트렉시드 (500 mg/m2) 그리고 시스플라틴 (75 mg/m2) 또는 카보플라틴 (AUC5) 중 연구자가 선택한 약물을 4주기 동안 21일 주기의 Day1에 정맥 내 투여한 후, 이 약 (80 mg) 1일 1회 경구 투여 및 페메트렉시드 (500 mg/m2) 3주마다 정맥 내 투여 (n=279)
‧ 이 약 (80 mg) 1일 1회 경구 투여 (n=278)
무작위 배정은 인종 (중국인/아시아인, 비중국인/아시아인 또는 비아시아인), WHO 수행 능력 (0 또는 1), 및 조직 검사 방법 (중앙 또는 현지)에 따라 층화되었다. 환자는 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더 이상 임상적 유익성을 경험하지 않을 때까지 시험 치료를 투여 받았다.
일차 유효성 평가변수는 RECIST 1.1에 따라 연구자가 평가한 PFS였다. 추가적인 유효성 평가변수에는 연구자의 평가에 따른 OS, ORR, DoR, PFS2, TFST 및 TSST가 포함되었다. BICR에 의해 평가된 CNS PFS, CNS ORR, CNS DoR 그리고 PRO도 평가되었다.
전체 시험 집단에 대한 베이스라인 인구통계학적 특성 및 질병 특성은 다음과 같았다: 연령 중앙값 61세 (범위 26‑85세), ≧75세 (8%), 여성 (61%), 아시아인 (64%), 백인 (28%), 흡연 미경험자 (66%). 모든 환자는 WHO 수행 능력이 0 또는 1이었다; 환자의 49%는 전이성 골 질환이 있었고, 환자의 53%는 흉곽 외 전이가 있었으며 20%는 간 전이가 있었다. 환자의 41%는 CNS 전이가 있었다 (베이스라인에 CNS 병변 부위, CNS 전이에 대한 병력, 및/또는 이전 수술, 및/또는 이전 방사선요법을 근거로 연구자가 확인함).
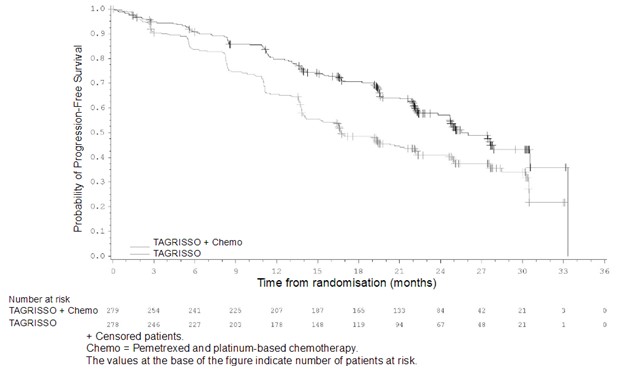
페메트렉시드 및 백금 기반 항암화학요법과 병용하는 이 약은 이 약의 단일요법과 비교하여 임상적으로 의미 있고 통계적으로 유의한 PFS 개선을 입증하였다 (51% 성숙; HR=0.62, 95% CI: 0.49, 0.79; P<0.0001; 중앙값 각각 25.5개월 및 16.7개월). OS 중간 분석 시점에, 페메트렉시드 및 백금 기반 항암화학요법과 병용한 이 약 투여 군의 환자들은 이 약의 단일요법 군에 비해 OS에 영향을 받지 않았다 (27% 성숙; HR=0.90, 95% CI: 0.65, 1.24; P = 0.5238).
연구자 평가에 따른 FLAURA2의 유효성 결과는 표 12에 요약되어 있으며, PFS에 대한 Kaplan‑Meier 곡선은 그림 9에 나타내었다.
표 12. 연구자 평가에 따른 FLAURA2에서의 유효성 결과
|
유효성 지표 |
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=279) |
이 약 (N=278) |
|
무진행 생존 기간(Progression-Free Survival) | ||
|
사건 수 (%) |
120 (43) |
166 (60) |
|
PFS 중앙값, 개월 (95% CI) |
25.5 (24.7, NC) |
16.7 (14.1, 21.3) |
|
HR (95% CI); P-값 |
0.62 (0.49, 0.79); P<0.0001 | |
|
전체 생존 기간(Overall Survival) | ||
|
사망 수 (%) |
71 (25) |
78 (28) |
|
OS 중앙값, 개월 (95% CI) |
NC (31.9, NC) |
NC (NC, NC) |
|
HR (95% CI); P-값 |
0.90 (0.65, 1.24); P=0.5238a | |
|
객관적 반응률(Objective Response Rate)b,c | ||
|
반응자 수 (n), 반응률 % (95% CI) |
232 83 (78, 87) |
210 76 (70, 80) |
|
오즈비 (95% CI); P-값d |
1.61 (1.06, 2.44); P=0.0261 | |
|
반응 기간 (Duration of Response, DoR)b | ||
|
DoR 중앙값, 개월 (95% CI)e |
24.0 (20.9, 27.8) |
15.3 (12.7, 19.4) |
|
질병 조절률 (Disease Control Rate) | ||
|
질병 조절되는 환자 수 (n), 조절률 % (95% CI) |
266 95 (92, 98) |
261 94 (90, 96) |
|
오즈비 (95% CI); P-값d |
1.33 (0.63, 2.81); P=0.4483 | |
|
첫 번째 후속 치료 시작 후 두 번째 PFS(PFS2) | ||
|
PFS2 사건 수 (%) |
81 (29) |
110 (40) |
|
PFS2 중앙값, 개월 (95% CI) |
30.6 (29.0, NC) |
27.8 (26.0, NC) |
|
HR (95% CI); P-값 |
0.70 (0.52, 0.93); P=0.0132 | |
|
무작위 배정부터 첫 번째 후속 치료 또는 사망까지의 시간(TFST) | ||
|
첫 번째 후속 치료를 받았거나 사망한 환자 수(%) |
104 (37) |
129 (46) |
|
TFST 중앙값, 개월 (95% CI) |
30.7 (27.3, NC) |
25.4 (22.8, NC) |
|
HR (95% CI); P-값 |
0.73 (0.56, 0.94); P=0.0159 | |
|
무작위 배정부터 두 번째 후속 치료 또는 사망까지의 시간(TSST) | ||
|
두 번째 후속 치료를 받았거나 사망한 환자 수(%) |
74 (27) |
103 (37) |
|
TSST 중앙값, 개월 (95% CI) |
NC (NC, NC) |
33.2 (28.2, NC) |
|
HR (95% CI); P-값 |
0.69 (0.51, 0.93); P=0.0157 | |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, ORR, DoR, DCR 결과는 RECIST 연구자 평가에 근거한 것이다.
PFS 추적관찰 시간의 중앙값은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 받은 환자에서 19.5개월이었고 이 약의 단독요법 군에서는 16.5개월이었다.
HR <1 및 오즈비 >1은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에 유리하다.
a 중간 분석(27% 성숙)에 따르면 통계적 유의성을 달성하려면 p‑값 < 0.00158 이 필요했다.
b 확인되지 않은 응답을 기반으로 했다.
c BICR에서 평가한 ORR 결과는 연구자 평가에서 보고된 것과 일치하였다; BICR 평가에 따른 ORR은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에서 92% (95% CI: 88, 95)였고 이 약의 단독요법 군에서 83% (95% CI: 78, 87)였다.
d 분석은 인종 (중국인/아시아인 vs. 비중국인/아시아인 vs. 비아시아인), WHO 수행 능력 (0 또는 1), 및 조직 검사 방법 (중앙 또는 현지)에 따라 계층화된 로지스틱 회귀 분석을 사용하여 수행되었다.
e Kaplan‑Meier 방법으로 계산했다.
그림 9. FLAURA2에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
이 약의 단일요법 대비 페메트렉시드 및 백금 기반 항암화학요법과 병용하는 이 약의 PFS 유익성은 민족성, 연령, 성별, 흡연 유무, 시험 시작 시 CNS 전이 상태, 및 EGFR 돌연변이 유형 (엑손 19 결손 또는 L858R)을 포함하여 사전에 정의된 모든 분석 하위군에 걸쳐 일관성 있게 관찰되었다.
베이스라인 대비 표적 병변 크기의 가장 우수한 백분율 변화 중앙값은 페메트렉시드 및 백금 기반 항암화학요법과 이 약 병용 투여 군에서 ‑53% (범위: 100%~20%)였고 이 약의 단일요법 군에서는 ‑50% (범위: ‑100%~40%)였다.
1차 치료로서 페메트렉시드 및 백금 기반 항암화학요법과 이 약 병용 투여 군에 무작위 배정된 환자도 이 약의 단일요법 군에 무작위 배정된 환자에 비해 임상적으로 의미있는 PFS2, TFST 및 TSST 개선을 보였다. 이러한 진행 후 평가변수 분석은 PFS 유익성이 후속 차수의 치료까지 대부분 유지되었음을 입증하였다.
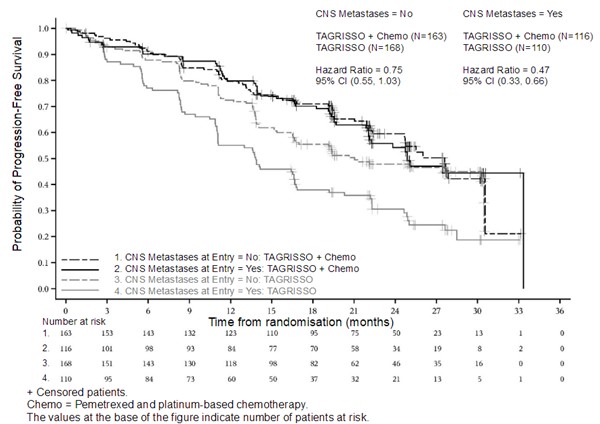
FLAURA2에서 CNS 전이 유효성 데이터
스테로이드가 필요하지 않은 무증상 CNS 전이 환자와 최종 치료 및 스테로이드 치료 완료 후 최소 2주 동안 신경학적 상태가 안정적인 환자는 FLAURA2 연구에 무작위 배정될 자격이 있었다. 모든 환자는 베이스라인 뇌 스캔이 가능했다. 수정된 RECIST를 사용한 BICR 평가에서는 CNS 측정 가능 및/또는 측정 불가능 병변(cFAS)이 있는 환자 222/557명(40%)의 하위 그룹과 CNS 측정 가능 병변(cEFR)이 있는 환자 78/557 (14%) 의 하위 그룹이 추가로 생성되었다. FLAURA2에 대한 BICR의 CNS 효능 평가 관련하여, 이 약의 단독요법 군에 비해 페메트렉시드와 백금 기반 항암화학요법과 이 약의 병용 투여 군에서 cFAS 및 cEFR에 대한 CNS PFS에서 임상적으로 의미 있는 개선을 입증했다(cFAS: 27% 성숙도, HR= 0.58, 95% CI 0.33, 1.01, P=0.0548[명목] 및 cEFR: 37% 성숙도, HR=0.40, 95% CI 0.19, 0.84, P=0.0157[명목]).
CNS 효능 데이터는 표 13에 요약되었다.
표 13. FLAURA2에서 베이스라인 뇌 스캔에서 CNS 전이가 있는 환자에 대해 BICR 평가된 CNS 유효성
|
유효성 지표 |
CNS 측정가능 및/또는 측정 불가능 병변 (cFAS) |
CNS 측정가능 병변 (cEFR) | ||
|
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=118) |
이 약 (N=104) |
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=40) |
이 약 (N=38) | |
|
CNS 무진행 생존 기간(CNS PFS)a | ||||
|
사건 수 (%) |
28 (24) |
31 (30) |
11 (28) |
18 (47) |
|
CNS PFS 중앙값, 개월 (95% CI) |
30.2 (28.4, NC) |
27.6 (22.1, NC) |
NC (23.0, NC) |
17.3 (13.9, NC) |
|
HR (95% CI); P-값 |
0.58 (0.33, 1.01); P=0.0548 |
0.40 (0.19, 0.84); P=0.0157 | ||
|
CNS 진행 없이 12개월 째 생존 (%) (95% CI) |
87 (79, 92) |
83 (73, 89) |
89 (74, 96) |
73 (55, 85) |
|
CNS 진행 없이 24개월 째 생존 (%) (95% CI) |
74 (63, 82) |
54 (39, 67) |
65 (43, 80) |
37 (18, 57) |
|
CNS 객관적 반응률a | ||||
|
CNS 반응 수(n), CNS 반응률 % (95% CI) |
86 73 (64, 81) |
72 69 (59, 78) |
35 88 (73, 96) |
33 87 (72, 96) |
|
완전 관해 (Complete response) |
70 (59) |
45 (43) |
19 (48) |
6 (16) |
|
부분 관해 (Partial response) |
16 (14) |
27 (26) |
16 (40) |
27 (71) |
|
오즈비 (95% CI); P-값b,c |
1.19 (0.67, 2.14); P=0.5492 |
1.06 (0.28, 4.00); P=0.9308 | ||
|
CNS Duration of Responsea | ||||
|
CNS DoR 중앙값, 개월 (95% CI) |
NC (23.8, NC) |
26.2 (19.4, NC) |
NC (21.6, NC) |
20.9 (12.6, NC) |
|
12개월 시점에 반응 보이는 환자 (%) (95% CI) |
93 (85, 97) |
81 (68, 89) |
93 (75, 98) |
74 (53, 87) |
|
24개월 시점에 반응 보이는 환자 (%) (95% CI) |
62 (40, 77) |
57 (38, 72) |
57 (27, 78) |
45 (22, 65) |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
HR <1 및 오즈비 >1은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에 유리하다.
a 확인되지 않은 응답을 기반으로 했다.
b 명목(Nominal) P‑값
c 분석은 치료 요인을에 대한 로지스틱 회귀분석을 이용하여 수행했다.
베이스라인 대비 표적 CNS 병변 크기의 최고 백분율 변화 중앙값은 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서 ‑94%(범위: ‑100% ~ 7%)였고, 이 약의 단독요법 군에서 ‑61%(범위: ‑100% ~ 68%)이었다.
FLAURA2에서는 CNS 전이 상태(기준시 CNS 병변 부위, 병력 및/또는 이전 수술 및/또는 CNS 전이에 대한 이전 방사선 치료를 기반으로 연구자가 식별함)를 기반으로 사전 지정된 PFS 하위군으로 연구를 시작했다. 연구 시작 시 CNS 전이 상태와 관계없이, 이 약의 단독요법 군에 비해 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서의 PFS 개선이 입증되었다. 연구 시작 시 CNS 전이 상태별 PFS에 대한 Kaplan‑Meier 곡선을 그림 10에 제시했다.
그림 10. FLAURA2에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
환자 보고 증상 및 HRQL은 EORTC QLQ C30 및 그에 대한 폐암 모듈 (EORTC QLQ‑LC13)을 이용해 전자적으로 수집되었다. LC13은 첫 8주 동안 주1회 시행되었고, 이후 진행 전에는 3주마다 그리고 진행 후에는 8주마다 시행되었다. C30은 첫 8주 동안 3주마다 평가되었고, 이후 진행 전에는 6주마다 그리고 진행 후에는 8주마다 평가되었다. 베이스라인에, 환자 보고 증상, 신체 기능 또는 GHS/QoL에서 페메트렉시드 및 백금기반 화학요법과 병용하는 이 약 군과 이 약의 단일요법군 간에 차이는 관찰되지 않았다. 첫 19개월에 걸친 순응도는 전반적으로 높았고 (≧80%) 두 군 모두 유사했다.
주요 폐암 증상 분석
베이스라인부터 제19개월까지 수집된 자료는 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군과 이 약의 단일요법 군에서 사전 명시된 5가지 일차 PRO 증상 중 3가지 (기침, 호흡곤란, 및 흉통)에 대해 유사한 개선을 나타냈으며, 기침에서의 개선은 두 군 모두 확립된 임상적으로 관련된 기준치 (베이스라인 대비 변화 ≦‑10)에 도달했다. 이 약의 단일요법군에서는 식욕 상실 및 피로에 대해 개선되는 추세를 보였다. 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서는 화학요법의 첫 4주기 동안 피로가 악화되는 추세를 보이다가 이후 개선되는 추세를 보였으며, 식욕 상실은 악화되는 추세를 보였다. 이러한 변화는 임상적으로 의미가 없었다. 이러한 자료는 표 14에 제시되어 있다.
표 14. 혼합 모델 반복 측정 ‑ 주요 폐암 증상 – 이 약 단독 요법으로 치료받은 환자와 페메트렉시드 및 백금 기반 항암화학요법과 이 약을 병용하여 치료받은 환자의 베이스라인으로부터의 평균 변화
|
|
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=279) |
이 약 (N=278) |
치료에서의 예상되는 차이 (95%CI) | ||
|
N |
Adjusted mean |
N |
Adjusted mean | ||
|
기침 |
253 |
-13.23 |
251 |
-11.19 |
-2.04 (-4.35, 0.26) |
|
호흡 곤란 |
253 |
-3.09 |
251 |
-5.67 |
2.57 (0.28, 4.86) |
|
가슴 통증 |
253 |
-6.33 |
251 |
-6.61 |
0.29 (-1.62, 2.20) |
|
식욕 감퇴 |
253 |
2.87 |
253 |
-4.58 |
7.45 (4.52, 10.38) |
|
피로감 |
253 |
-0.03 |
253 |
-6.31 |
6.28 (3.60, 8.96) |
전반적인 건강상태/QoL 및 신체기능 개선 분석
두 군 모두 신체 기능 및 GHS/QoL에 임상적으로 의미 있는 변화가 없다고 보고했다.
이전에 치료 받은 적 있는 T790M양성 비소세포폐암 환자 ‑ AURA3
TKI 치료 후 또는 치료 중 진행되면서 국소진행 또는 전이된 T790M 비소세포폐암 환자 치료 시 이 약의 유효성 및 안전성은 무작위 배정된, 공개, 활성 대조군 임상 3상시험(AURA 3)에서 증명되었다.
환자들은 이 약 (n=279) 또는 백금기반 이중 항암화학요법제(n=140)에 2:1 (이 약: 백금기반 이중 항암화학요법제) 비율로 무작위 배정되었다. 무작위 배정은 민족성 (아시아인 및 비아시아인)에 따라 층화되었다. 이 약 군의 환자는 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더이상 임상적 유익성을 경험하지 않을 때까지 이 약 80 mg을 1일 1회 경구 투여 받았다.
화학요법은 최대 6주기동안 매 21일 주기의 제 1일째에 페메트렉시드 500mg/m2와 카보플라틴 AUC5 병용요법 또는 페메트렉시드 500mg/m2와 시스플라틴 75mg/m2병용요법으로 이루어졌다. 4주기의 백금기반 화학요법 후에 질병이 진행되지 않은 환자는 페메트렉시드 유지요법을 받을 수 있었다 (매 21일 주기의 제 1일째에 페메트렉시드 500mg/m2). 화학요법군의 시험대상자 중 객관적인 방사선학적 진행이 있는 환자 (연구자에 의해 그리고 독립적인 중앙 영상 검토에 의해 확정된)는 이 약으로 치료를 시작할 수 있는 기회가 주어졌다.
AURA 3시험에서 항암화학요법군 대비 이 약으로 치료받은 환자군에서 통계적으로 유의한 PFS 개선이 증명되었다. AURA 3 연구자 평가에 따른 AURA 3의 유효성 결과는 표 15에 요약되어 있고, 그림 11에 PFS에 대한 Kaplan‑Meier 곡선을 나타내었다. 최종 OS 분석 (67 % 성숙 시 실시)에서 치료군 간에 통계적으로 유의한 차이가 관찰되지 않았으며, 이 시점에는 항암화학요법에 무작위 배정된 99명의 환자 (71 %)가 이 약 치료로 넘어가 있었다.
표 15. 연구자 평가에 따른 AURA3에서의 유효성 결과
|
유효성 지표 |
이 약(N=279) |
항암화학요법 (N=140) |
|
무진행생존기간(Progression-Free Survival) | ||
|
사건 수 (% maturity) |
140 (50) |
110 (79) |
|
PFS 중앙값, 개월 (95% CI) |
10.1 (8.3, 12.3) |
4.4 (4.2, 5.6) |
|
HR (95% CI) ; P-값 |
0.30 (0.23,0.41); P <0.001 | |
|
전체 생존기간(Overall Survival, OS)1 | ||
|
사망 수 (% maturity) |
188 (67.4) |
93 (66.4) |
|
OS 중앙값, 개월 (95% CI) |
26.8 (23.5, 31.5) |
22.5 (20.2, 28.8) |
|
HR (95.56% CI); P-값 |
0.87 (0.67, 1.13); P=0.277 | |
|
객관적 반응률(Objective Response Rate)2 | ||
|
반응자 수, 반응률 (95% CI) |
197 71% (65, 76) |
44 31% (24, 40) |
|
오즈비 (95% CI) ; P-값 |
5.4 (3.5, 8.5); P <0.001 | |
|
반응 기간(Duration of Response, DoR)2 | ||
|
DoR 중앙값, 개월 (95% CI) |
9.7 (8.3, 11.6) |
4.1 (3.0, 5.6) |
HR=위험비; CI=신뢰구간; OS=전체 생존기간
RECIST 연구자 평가에 근거한 모든 유효성 결과
HR <1 및 오즈비 >1은 이 약에 유리하다.
1 OS의 최종 분석은 67% 성숙 시 실시되었다. HR에 대한 CI는 이전 중간 분석을 위해 조정되었다. OS 분석은 교차에 대한 잠재적인 교란혼재효과는 보정되지 않았다(항암화학요법군의 99명 [71%] 환자는 이후 오시머티닙 치료를 받았다.).
2 연구자 평가에 의한 ORR 및 DoR 결과는 눈가림된 독립적 중앙 검토(Blinded Independent Central Review, BICR)를 통해 보고된 것과 일치한다; BICR 평가에 의한 ORR은 오시머티닙 치료 시 64.9% [95% CI: 59.0, 70.5], 항암화학요법제 치료 시 34.3% [95% CI: 26.5, 42.8]였다. ; BICR에 의한 DoR은 오시머티닙 치료 시 11.2개월(95% CI: 8.3, NC), 항암화학요법제 치료 시 3.1 개월(95% CI: 2.9, 4.3)이었다.
그림 11. AURA3에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
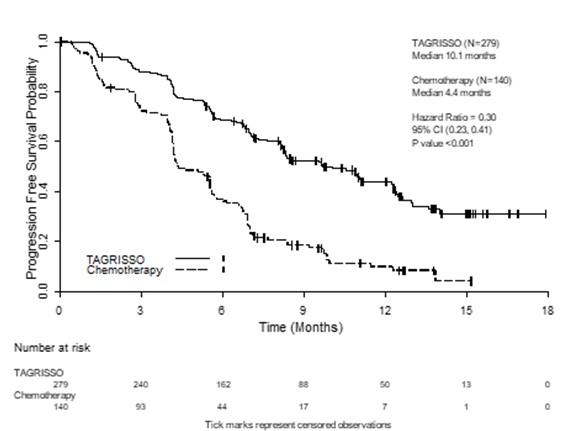
항암화학요법제를 투여 받은 환자군과 비교하여 이 약을 투여 받은 환자군에게 유리한 0.50 미만 HR을 보이는 임상적으로 의미 있는 PFS 개선은 민족성, 나이, 성별, 흡연 유무, 시험 시작 시 CNS 전이 상태, EGFR 변이 (엑손 19 결손 또는 L858R), 및 EGFR‑TKI 1차 치료 기간을 포함하여 사전에 정의된 모든 분석 하위군에서 일관성 있게 관찰되었다.
4) 독성시험 정보
반복 투여 독성
랫드와 개에 대한 반복 투여 독성시험에서 관찰된 주요 결과는 눈(각막), 위장관(혀 포함), 피부 및 수컷과 암컷 생식기관의 상피에 영향을 미치는 위축성, 염증성 그리고/또는 퇴행성 변화로 구성되었다. 이러한 결과는 80 mg 치료 용량의 환자들에서 관찰된 것보다 낮은 혈장 농도에서 발생하였다. 투여 1개월 후 발생한 결과는 투여 중단 후 1개월 이내에 대체로 회복을 나타내었다.
발암성 및 돌연변이성
오시머티닙은 26주 동안 Tg rasH2 유전자 이식 마우스에게 경구 투여했을 때 발암 가능성을 보이지 않았다.
랫드의 104주 발암성 연구에서 1일 1회 80 mg의 권장 임상 용량에서 관찰된 AUC의 0.2배 노출에서 수정체 섬유 변성 및 장간막 림프절에서 증식성 혈관 병변 (혈관종 과형성 및 혈관종)의 발생률 증가가 관찰되었다. 수정체 섬유 변성은 52주차에 처음 관찰되고 투여 기간이 증가함에 따라 발생률 및 중증도가 점진적으로 증가하는 수정체 혼탁에 대한 검안경 관찰과 일치했다. 이러한 결과의 임상적 연관성은 배제할 수 없다. 장간막 림프절에서의 증식성 혈관 병변(혈관종 과형성 및 혈관종)은 인간의 혈관 신생물에 대한 발암 가능성을 예측하는 것은 아니다.
오시머티닙은 in vitro 및 in vivo 시험에서 유전적 손상을 유발하지 않았다.
생식 독성
동물에 대한 시험에 근거하여 수컷 수태능은 이 약 치료로 손상될 수 있다. 1개월 이상 오시머티닙에 노출된 랫드와 개에서 퇴행성 변화를 고환에서 보였으며, 3개월 동안 오시머티닙에 노출된 후 랫드에서 수컷 수태능이 감소하였다. 이러한 결과들은 임상적으로 관련된 혈장 농도에서 관찰되었다. 1개월 투여 후 보인 고환에서의 병리학적 소견은 랫드에서 가역적이었으나, 개에서 이러한 병변의 가역성에 대한 확정적인 진술은 할 수 없다.
동물에서의 시험에 근거하여 암컷 수태능은 이 약 치료로 손상될 수 있다. 반복투여 독성시험에서 임상적으로 관련된 혈장 농도로 오시머티닙에 1개월 이상 노출된 랫드에서 발정휴지기 증가, 난소에서 황체 퇴행 및 자궁과 질의 상피 얇아짐이 관찰되었다. 1개월 투여 후 관찰된 난소에서의 결과는 가역적이었다. 랫드에 대한 암컷 수태능 시험에서, 오시머티닙 20 mg/kg/day 투여 (권장 1일 임상 용량인 80 mg과 거의 동등한 용량)는 발정 주기 또는 임신하는 암컷의 수에 영향이 없었으나, 초기 배아 사망을 야기했다. 이러한 결과는 1개월간 휴약 후 가역성의 증거를 보였다.
랫드에 대한 수정된 배태자 발달 시험에서, 오시머티닙은 임신한 랫드에게 배아 착상 전 투여 시 배아 치사를 초래했다. 이러한 영향은 권장 용량 1일 80 mg에서의 사람 노출과 동등한 노출 (총 AUC에 근거)인 20 mg/kg/day의 모체 내약 용량 (maternally tolerated dose)에서 관찰되었다. 기관 형성기에 20 mg/kg 이상의 용량에 대한 노출은 태자 무게 감소를 유발하였으나, 태자의 외형 또는 내장 형태에는 유해한 영향을 미치지 않았다. 오시머티닙을 임신한 암컷 랫드에게 임신 기간에 걸쳐서, 그리고 이후 수유 초기에 투여했을 때, 수유를 받는 차산자에서 오시머티닙 및 그 대사체에 대한 노출을 보였으며, 차산자의 생존 감소 및 차산자 성장 감소가 있었다. (20 mg/kg 이상의 용량에서)
단독요법
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 비소세포폐암 환자에서 완전 종양 절제술 후 보조 치료
◦ EGFR 엑손 19 결손 또는 엑손 21 (L858R) 치환 변이가 있고 백금 ‑기반 화학 방사선 요법 중 또는 후 질병이 진행되지 않은 절제 불가능한 국소 진행성(III기) 비소세포폐암 환자의 치료
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 국소 진행성 또는 전이성 비소세포폐암 환자의 1차 치료
▪ 이전에 EGFR ‑TKI로 치료 받은 적이 있는 EGFR T790M 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자의 치료
병용요법
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 국소 진행성 또는 전이성 비편평 비소세포폐암 환자의 1차 치료에서 페메트렉시드와 백금 기반 항암화학요법과 병용 요법
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 비소세포폐암 환자에서 완전 종양 절제술 후 보조 치료
◦ EGFR 엑손 19 결손 또는 엑손 21 (L858R) 치환 변이가 있고 백금 ‑기반 화학 방사선 요법 중 또는 후 질병이 진행되지 않은 절제 불가능한 국소 진행성(III기) 비소세포폐암 환자의 치료
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 국소 진행성 또는 전이성 비소세포폐암 환자의 1차 치료
▪ 이전에 EGFR ‑TKI로 치료 받은 적이 있는 EGFR T790M 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자의 치료
병용요법
▪ EGFR 엑손 19 결손 또는 엑손 21(L858R) 치환 변이된 국소 진행성 또는 전이성 비편평 비소세포폐암 환자의 1차 치료에서 페메트렉시드와 백금 기반 항암화학요법과 병용 요법
이 약을 투여하는 경우, 치료 시작 전에 EGFR 변이 상태를 평가해야 한다. 다음에 대해 충분히 검증된 신뢰성 있는 시험방법을 사용하여 확인하여야 한다.
‑ 보조 치료, 절제 불가능한 국소 진행성 종양 치료 및 1차 치료: 엑손19 결손 또는 엑손21(L858R) 치환 변이
‑ 이전에 EGFR ‑TKI로 치료 받은 적이 있는 환자: T790M 변이; 식품의약품안전처에서 동 의약품의 사용에 적합하게 허가된 체외진단용 의료기기를 사용해야 함
단독요법
이 약의 권장 용량은 1일 1회 오시머티닙 80 mg이다.
병용요법
페메트렉시드와 백금 기반 항암화학요법과 병용할 때 이 약의 권장 용량은 1일 1회 오시머티닙 80 mg이다.
페메트렉시드와 시스플라틴 또는 카보플라틴 각각에 대한 투여량 정보는 해당 약물의 허가사항을 참조한다(13. 전문가를 위한 정보 참조).
이 약은 매일 일정한 시간에 식사와 관계없이 복용한다.
치료 기간
보조 치료 환자는 질병이 재발하거나 수용할 수 없는 독성이 나타날 때까지 복용을 지속한다. 3년을 초과하는 치료 기간은 연구되지 않았다.
국소 진행성 또는 전이성 폐암 환자는 질병이 진행되거나 허용할 수 없는 독성이 나타날 때까지 복용을 지속한다.
투여방법
이 약은 경구투여하며 물과 함께 통째로 삼켜야 한다. 정제를 부수거나 쪼개거나 씹어서는 안 된다.
환자가 정제를 삼킬 수 없는 경우에는, 우선 비탄산수 50 mL에 녹인다. 정제를 부수지 않고 물에 넣고 녹을 때까지 저은 후 즉시 마신다. 그 다음, 잔류물이 남지 않도록 물 약 100 mL을 추가하여 즉시 마신다. 다른 액체를 추가하지 않도록 한다.
비위관(nasogastric tube)을 통한 투여가 필요한 경우, 위와 동일한 과정을 따르되 처음 녹일 때 물 15 mL를 사용하고 잔류물을 헹구는데 15 mL를 사용한다.
약물을 녹인 용액과 잔류물을 헹군 용액은 정제를 물에 넣은 지 30분 안에 투여되어야 한다.
투여 누락
이 약의 복용을 누락한 경우, 다음 투여까지 12시간 이상 남았으면 즉시 복용한다.
용량 조절
각 환자의 안전성 및 내약성에 근거하여 투여 중단 및/또는 용량 감소가 필요할 수 있다. 용량 감소가 필요한 경우, 이 약의 용량은 1일 1회 40 mg으로 감량되어야 한다. 이상반응 독성에 대한 용량 감소 가이드라인은 표 1에 제시되어 있다.
표 1. 권장 용량 조절
<SUP>a</SUP>비고: 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0에 의해 등급이 분류된 임상적 이상반응의 중증도.
<SUP>b </SUP>사용상의 주의사항 1. 경고 참조.
병용요법: 이 약을 페메트렉시드 및 백금 기반 항암화학요법과 병용투여 시 이상반응이 발생한 경우, 투여 약물 중 어느 하나의 투여를 적절히 조절해야 한다. 이 약의 용량 조절은 표 1을 참고한다. 페메트렉시드, 시스플라틴 또는 카보플라틴은 각각의 허가사항에 따라 투여 중지, 용량 감소, 또는 영구 중단해야 한다.
‑ 보조 치료, 절제 불가능한 국소 진행성 종양 치료 및 1차 치료: 엑손19 결손 또는 엑손21(L858R) 치환 변이
‑ 이전에 EGFR ‑TKI로 치료 받은 적이 있는 환자: T790M 변이; 식품의약품안전처에서 동 의약품의 사용에 적합하게 허가된 체외진단용 의료기기를 사용해야 함
단독요법
이 약의 권장 용량은 1일 1회 오시머티닙 80 mg이다.
병용요법
페메트렉시드와 백금 기반 항암화학요법과 병용할 때 이 약의 권장 용량은 1일 1회 오시머티닙 80 mg이다.
페메트렉시드와 시스플라틴 또는 카보플라틴 각각에 대한 투여량 정보는 해당 약물의 허가사항을 참조한다(13. 전문가를 위한 정보 참조).
이 약은 매일 일정한 시간에 식사와 관계없이 복용한다.
치료 기간
보조 치료 환자는 질병이 재발하거나 수용할 수 없는 독성이 나타날 때까지 복용을 지속한다. 3년을 초과하는 치료 기간은 연구되지 않았다.
국소 진행성 또는 전이성 폐암 환자는 질병이 진행되거나 허용할 수 없는 독성이 나타날 때까지 복용을 지속한다.
투여방법
이 약은 경구투여하며 물과 함께 통째로 삼켜야 한다. 정제를 부수거나 쪼개거나 씹어서는 안 된다.
환자가 정제를 삼킬 수 없는 경우에는, 우선 비탄산수 50 mL에 녹인다. 정제를 부수지 않고 물에 넣고 녹을 때까지 저은 후 즉시 마신다. 그 다음, 잔류물이 남지 않도록 물 약 100 mL을 추가하여 즉시 마신다. 다른 액체를 추가하지 않도록 한다.
비위관(nasogastric tube)을 통한 투여가 필요한 경우, 위와 동일한 과정을 따르되 처음 녹일 때 물 15 mL를 사용하고 잔류물을 헹구는데 15 mL를 사용한다.
약물을 녹인 용액과 잔류물을 헹군 용액은 정제를 물에 넣은 지 30분 안에 투여되어야 한다.
투여 누락
이 약의 복용을 누락한 경우, 다음 투여까지 12시간 이상 남았으면 즉시 복용한다.
용량 조절
각 환자의 안전성 및 내약성에 근거하여 투여 중단 및/또는 용량 감소가 필요할 수 있다. 용량 감소가 필요한 경우, 이 약의 용량은 1일 1회 40 mg으로 감량되어야 한다. 이상반응 독성에 대한 용량 감소 가이드라인은 표 1에 제시되어 있다.
표 1. 권장 용량 조절
|
표적 기관 |
이상반응a |
용량 변경 |
|
폐b |
간질성 폐질환(ILD)/폐염증 |
이 약을 영구 중단한다. |
|
확정적 백금-기반 화학 방사선 요법 후ILD/폐염증: 무증상(1등급) |
적절히 이 약을 지속하거나 일시 중지 및 재개한다. | |
|
확정적 백금-기반 화학 방사선 요법 후ILD/폐염증: 2등급 이상 |
이 약을 영구 중단한다. | |
|
심장b |
최소 2회의 별도의 ECG에서 500 msec 초과의 QTc 간격 |
QTc 간격이 481msec 미만이 될 때까지, 또는 베이스라인 QTc가 481msec 이상인 경우 베이스라인으로 회복될 때까지 이 약을 중단하고, 이후에 감량된 용량(40mg)으로 다시 시작한다. |
|
중대한 부정맥 징후/증상이 동반된 QTc 간격 연장 |
이 약을 영구 중단한다. | |
|
무증상성, 좌심실 박출률 기저치 대비 10 퍼센트포인트 이상 감소 및 50% 미만 |
최대 4주 동안 이 약 투여를 중지한다. 기저치로 돌아온다면 투여를 다시 시작한다. 기저치로 되돌아오지 않는다면 이 약을 영구 중단한다. | |
|
증상성 울혈성 심부전 |
이 약을 영구 중단한다. | |
|
피부b |
스티븐스-존슨증후군 및 독성 표피 괴사 용해 |
이 약을 영구 중단한다. |
|
혈액 및 림프계b |
재생 불량성 빈혈 |
이 약을 영구 중단한다. |
|
기타 |
3등급 이상의 이상반응 |
최대 3주간 이 약을 중단한다. |
|
3등급 이상의 이상반응이 이 약을 최대 3주간 중단한 이후 0-2등급으로 개선되는 경우 |
이 약을 동일 용량(80 mg) 또는 저용량(40 mg)으로 다시 시작 할 수 있다. | |
|
최대 3주간 이 약을 중단한 이후 0-2등급으로 개선되지 않는 3등급 이상의 이상반응 |
이 약을 영구 중단한다. |
<SUP>b </SUP>사용상의 주의사항 1. 경고 참조.
병용요법: 이 약을 페메트렉시드 및 백금 기반 항암화학요법과 병용투여 시 이상반응이 발생한 경우, 투여 약물 중 어느 하나의 투여를 적절히 조절해야 한다. 이 약의 용량 조절은 표 1을 참고한다. 페메트렉시드, 시스플라틴 또는 카보플라틴은 각각의 허가사항에 따라 투여 중지, 용량 감소, 또는 영구 중단해야 한다.
1. 경고
1) 간질성 폐질환(Interstitial Lung Disease, ILD)
ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들에서 이 약을 단독요법으로 투여받은 1813명의 환자 중 간질성 폐질환 또는 간질성 폐질환 유사 이상반응(예. 폐염증)은 4.0%의 환자에게서 보고되었으며, 이 중 치명적인 건은 0.4%(n=7) 보고되었다. 간질성 폐질환은 일본인 환자의 11.2%, 일본인이 아닌 아시아인 환자의 2.3%, 비아시아인 환자의 2.7%에게서 발생하였다. 첫 번째 투여부터 간질성 폐질환 또는 간질성 폐질환 유사 이상반응들의 발현시점의 중앙값은 2.8개월이었다.
FLAURA2에서 페메트렉시드 및 백금 기반 항암화학요법과 병용하여 이 약을 투여받은 276명의 환자 중 간질성 폐질환 또는 간질성 폐질환 유사 이상반응은 3.3%에서 보고되었으며, 이 중 치명적인 건은 0.4% (n=1)이었다. 간질성 폐질환은 일본인 환자의 14.9% 및 비아시아인 환자의 1.7%에게서 발생하였다. FLAURA2 병용요법 군에서 일본인이 아닌 아시아인 환자에서는 간질성 폐질환이 발생하지 않았다. 첫 번째 투여부터 간질성 폐질환 또는 간질성 폐질환 유사 이상반응들의 발현시점의 중앙값은 5.3개월이었다.
간질성 폐질환을 시사할 수 있는 호흡기 증상(예: 호흡곤란, 기침, 발열)의 악화가 나타나는 환자는 이 약을 중단하고 즉시 간질성 폐질환에 대해 조사한다. 간질성 폐질환이 확인되면 이 약을 영구 중단한다.
확정적 백금‑기반 화학 방사선 요법 후 간질성 폐질환
LAURA 임상시험에서 확정적 백금‑기반 화학 방사선 요법 후, 이 약을 투여받은 143명의 환자 중 56%(80명)와 위약을 투여받은 73명의 환자 중 38%(28명)에서 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응(예: 폐염증)이 보고되었다. 이 약 군에서는 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응 중 0.7%(1명)의 치명적 증례가 보고되었으며, 3등급은 3.5%, 2등급은 34%, 1등급은 18%이었다. 이 약을 투여받은 환자 중 7%가 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응으로 투여를 영구 중단 했으며, 35%는 일시 중단했다. 재투여를 받은 환자 46명 중 11%는 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 재발하였다. 이 약을 투여받고 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생한 80명의 환자 중 40%는 회복, 1.3%는 후유증을 동반하고 회복, 16%는 회복 중이었으며, 41%는 회복되지 않고 1.3%(1명)는 사망하였다.
확정적 백금‑기반 화학 방사선 요법 후 이 약으로 치료받은 환자에서 간질성 폐질환을 시사할 수 있는 호흡기 증상(예: 호흡곤란, 기침, 발열)의 악화가 나타나는 환자는 이 약을 중단하고 즉시 간질성 폐질환에 대해 조사한다. 간질성 폐질환이 확인되면 무증상(1등급) 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생하는 경우, 적절히 이 약을 지속하거나 일시 중지 및 재개한다. 2등급 이상의 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생하는 경우, 이 약의 사용을 영구 중단한다.
2) 스티븐스‑존슨증후군, 다형성 홍반 (Erythema multiforme) 및 독성 표피 괴사 용해
이 약 치료와 관련하여, 다형성 홍반 및 독성 표피 괴사 용해의 사례 보고는 흔하지 않게 보고되었고, 스티븐스‑존슨증후군은 드물게 보고되었다. 치료를 시작하기 전에 환자에게 스티븐스‑존슨증후군, 다형성 홍반 및 독성 표피 괴사 용해의 징후와 증상을 알려야 한다.
스티븐스‑존슨증후군 또는 독성 표피 괴사 용해를 연상시키는 징후와 증상이 나타나면, 이 약을 일시 중단해야 한다. 스티븐스‑존슨증후군 또는 독성 표피 괴사 용해가 진단되면, 이 약을 즉시 중지해야 한다.
다형성 홍반을 연상시키는 징후와 증상이 있는 경우, 긴밀한 환자 모니터링 및 이 약의 중단 또는 중지를 고려해야 한다.
3) QTc 간격 연장
이 약 단독요법 (80mg)으로 치료한 ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들의 1813명 환자 중, 1.1%(n=20)는 500 msec 초과 QTc를, 4.3%(n=78)는 베이스라인 QTc로부터 60 msec를 초과하는 QTc 증가를 나타냈다. 이 약에 대한 약동학적 분석 시 QTc 간격 연장의 농도 의존적 증가가 예측되었다. ADAURA, LAURA, FLAURA, FLAURA2 또는 AURA 임상시험들에서 QTc와 관련된 부정맥 사례는 보고되지 않았다.
가능한 경우, 선천적으로 긴 QT 증후군이 있는 환자는 이 약의 사용을 피한다 (3.이상반응 참조). 울혈성 심부전, 전해질 이상이 있는 환자, 또는 QTc 간격이 연장되는 것으로 알려진 약물을 투여 중인 환자에서는 심전도(ECG) 및 전해질의 주기적인 모니터링을 고려한다. 심전도 검사에서 QTc 간격이 500 msec을 초과하는 결과가 적어도 2회 별도로 발생한 환자에서는 QTc 간격이 481msec 미만이 될 때까지, 또는 베이스라인 QTc가 481msec 이상인 경우 베이스라인으로 회복될 때까지 이 약을 중단하고, 이후에 용법용량 항 표 1에 기술된 바와 같이 감소된 용량으로 투여를 재개한다. 다음 중 어느 하나와 더불어 QTc 간격 연장이 발생한 환자는 이 약을 영구 중단한다: 염전성 심실빈맥(Torsade de pointes), 다형성 심실빈맥(polymorphic ventricular tachycardia), 중대한 부정맥의 징후/증상.
4) 심수축성/심장독성 변화
임상시험들에서 이 약을 투여받은 1813명의 환자 중 3.8%에서 심장독성(심부전, 만성 심부전, 울혈성 심부전, 폐부종, 박출률 감소)을 나타냈다. 이 중 1명(0.1%)은 치명적인 결과로 이어졌다.
임상시험에서 베이스라인 및 최소 1회의 좌심실박출률 추적 평가를 받은 이 약의 단독요법으로 치료받은 환자의 4.2%(65/1557)에서 좌심실 박출량이 10퍼센트포인트 이상 감소하고 50% 미만으로 나타났다. 위약 대조 시험(ADAURA)에서 이 약을 투여 받은 환자의 1.5%(5/325)와 위약을 투여 받은 환자의 1.5%(5/331)에서 10%포인트 이상 50% 미만의 좌심실박출률 감소를 경험했다. LAURA 임상시험에서 백금‑기반 화학 방사선 요법 후, 이 약으로 치료받은 환자의 3.0% (4/135명)와 위약으로 치료받은 환자 0명에서 좌심실 박출량이 10퍼센트포인트 이상 감소하고 50% 미만으로 나타났다. FLAURA2에서 베이스라인 및 최소 1회의 좌심실박출률 추적 평가를 받은 이 약과 페메트렉시드, 백금 기반 항암화학요법을 병용하여 치료받은 환자의 8.0%(21/262)에서 좌심실 박출량이 10%포인트 이상 감소하고 50% 미만으로 나타났다.
심장 위험인자가 있거나 좌심실박출률에 영향을 미칠 수 있는 상태의 환자에게는 베이스라인 및 치료 중 좌심실박출률 평가를 포함하는 심장 모니터링을 한다. 치료 중 관련된 심장 징후/증상이 있는 환자의 경우, 좌심실박출률 평가를 포함한 심장 모니터링을 한다. 좌심실박출률이 기저치 대비 10 퍼센트포인트 이상 감소하고 50% 미만으로 나타날 경우, 이 약 투여를 최대 4주간 중단하고 중단 후에도 증상이 개선되지 않을 때에는 이 약 투여를 영구 중단한다. 증상성 울혈성 심부전 발생 시에는 이 약의 투여를 영구 중단한다.
5) 각막염
ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들에서 이 약의 단독요법으로 치료된 1813명의 환자 중 0.6%(n=10)에서 각막염이 보고되었다. 다음과 같은 각막염의 징후 및 증상을 보이는 환자들은 즉시 안과전문의에게 상담을 받아야 한다: 눈의 염증, 눈물 분비, 빛에 민감한 눈, 시야 흐림, 눈의 통증 그리고/또는 눈의 충혈 (용법용량 항 참조)
6) 재생 불량성 빈혈
재생 불량성 빈혈이 이 약의 치료와 관련하여 드물게 보고되었다. 일부 사례는 치명적인 결과가 있었다. 치료를 시작하기 전에 환자에게 지속적인 발열, 멍, 출혈, 창백을 포함하되 이에 국한되지 않는 재생 불량성 빈혈의 징후와 증상에 대해 알려야 한다. 재생 불량성 빈혈을 암시하는 징후 및 증상이 발생하면 환자를 면밀히 모니터링하고 이 약의 중지 또는 중단을 고려해야 한다. 재생 불량성 빈혈이 확인된 환자는 이 약을 중단해야 한다.
2. 다음 환자에는 투여하지 말 것.
이 약의 주성분이나 부형제에 대해 중증의 과민증이 있는 환자
3. 이상반응
1) 안전성 프로파일의 전반적인 요약
EGFR 변이 양성 비소세포폐암 환자에 대한 시험
이 약 단독요법의 안전성은 EGFR 변이 양성 비소세포폐암 환자 1813명으로부터 수집된 데이터를 기반으로 한다. 이러한 환자들은 4건의 무작위배정된 3상 임상시험(ADAURA: 보조 치료; FLAURA 및 FLAURA2(단독요법 군):1차 치료; AURA3: 2차 치료), 2건의 단일군 2상 임상시험(AURAex 및 AURA2: 2차 치료 이상) 및 1건의 1상 임상시험(AURA1: 1차 치료 이상)에서 이 약 1일 80 mg을 투여 받았다.
대부분의 이상반응 중증도는 1등급 또는 2등급이었다. 가장 흔한 빈도로 보고된 약물이상반응은 설사(47%), 발진(46%), 손발톱주위염(34%), 건성 피부(32%), 및 구내염(24%)이었다. 이 약에서 나타난 3등급 및 4등급 이상반응은 각각 9.2%와 0.2%였다. 이 약 80 mg을 1일 1회 치료받은 환자들에게서, 약물이상반응으로 인한 용량감소는 3.6%의 환자들에게서 나타났다. 이상반응으로 인한 투약중단은 4.7%이었다.
백금‑기반 화학 방사선 요법 후 이 약(1일 1회 80 mg)의 안전성은 EGFR 돌연변이 양성 비소세포폐암 환자 143명에서 얻은 자료를 기반으로 한다.
페메트렉시드와 백금 기반 항암화학요법을 병용하여 투여한 이 약의 안전성은 EGFR 변이 양성 비소세포폐암 환자 276명 데이터를 기반으로 하며, 이 약의 단독요법 및 페멕트렉시드와 백금 기반 항암화학요법의 알려진 안전성 프로파일과 일치했다.
스테로이드 치료가 필요한 간질성 폐질환, 약물 유도 간질성 폐질환, 방사선 폐염증의 과거 병력이 있거나 임상적으로 활동성 간질성 폐질환 증거가 있는 환자는 임상시험에서 제외되었다. 안정 시 ECG (예를 들어, QTc 간격이 470 ms 초과)로 측정 시 리듬 및 전도가 임상적으로 중요한 비정상인 환자는 이러한 시험들에서 제외되었다. 스크리닝 및 이후 12주마다 환자의 좌심실박출률이 평가되었다.
AURAex 및 AURA2에 참여한 한국인 환자(66명)에서 전체 및 3/4등급 이상반응 발생율은 전체 환자군과 유사하였다. 다만, 한국인 환자에서 전체 환자군에 비해 상대적으로 많이 발생하는 이상반응은 발진, 소양증, 손발톱주위염, 혈소판 감소증, 백혈구 감소증으로 나타났다. 상대적으로 낮은 빈도로 발생하는 이상반응은 설사, 오심으로 보고되었다. 이들 이상반응은 대부분 1등급 또는 2등급이었다.
2) 이상반응 표
이상반응은 ADAURA, FLAURA, FLAURA2, AURA3, AURAex, AURA2, 및 AURA1 시험들에서 이 약의 단독요법으로 1일 80 mg을 투여 받은 EGFR 변이 양성 환자 1813명의 통합 데이터세트에서의 비교 가능한 이상반응 발생률에 근거하여 표 2에 빈도 카테고리로 분류되었다.
약물이상반응은 MedDRA 기관계 분류(System Organ Class;SOC)에 따라 나열하였다. 각 기관계 분류 내에서, 약물이상반응은 빈도 내림차순으로(즉, 가장 빈번한 반응이 제일 먼저 오도록) 나열하였다. 각 빈도 군 내에서, 약물이상반응은 중대성이 감소하는 순으로 나열하였다. 또한, 각 약물이상반응의 해당 빈도 범주는 CIOMS III 분류에 근거하여 다음과 같이 정의한다: 매우 흔하게(≧1/10), 흔하게(≧1/100 ~ <1/10), 흔하지 않게(≧1/1,000 ~ <1/100), 드물게(≧1/10,000 ~ <1/1000), 매우 드물게(<1/10,000), 빈도불명(사용 가능한 데이터로부터 추정할 수 없음).
표 2. ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 시험들에서 보고된 이상반응<SUP>a</SUP>
a ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 시험들(AURA3, AURAex, AURA2 및 AURA1)로부터의 데이터가 통합되었다. 무작위배정된 치료로서 이 약을 최소 1회 이상 투여 받은 환자의 사례만 요약하였다.
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란, 각막내피결손, 각막염, 점상각막염 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질성 폐렴 포함.
e 7 건의 CTCAE 5등급(치명적)이 보고됨.
f 입 궤양 형성, 구내염 포함
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성 발진(rash vesicular), 피부미란(skin erosion) 포함.
h 손발톱바닥장애(nail bed disorder), 손발톱바닥감염(nail bed infection), 손발톱바닥염증(nail bed inflammation), 손발톱변색(nail discolouration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k 시판 후 조사에서 지속색소이상홍반 사례가 보고되었다.
l ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 시험의 1813명 환자 중 6명이 다형성 홍반을 보고하였다. 시판 후 조사 연구(N=3578)로부터의 7건을 포함하여, 다형성 홍반의 시판 후 보고도 있었다.
m 추정 빈도. 추정치에 대한 95% CI의 상한은 3/1813 (0.17%)이다. 임상시험에서 보고 없음.
n 시판 후 연구에서 보고된 한 건의 사례이며, 빈도는 ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 연구와 시판 후 연구(N=5391)에서 도출되었다.
o QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
p 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 3. ADAURA<SUP>a</SUP>시험에서 보고된 이상반응
ADAURA 시험에서 시험 치료기간의 중앙값은 이 약 군의 환자에서 22.5 개월이었고 위약 군의 환자에서 18.7개월이었다.
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0
c 모든 사례는 3등급이었다. 사망은 없었다.
d 각막염(keratitis), 점상각막염(punctate keratitis), 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect) 포함.
e 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
f 구내염(stomatitis), 입 궤양 형성(mouth ulceration) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis), 전신 소양증(pruritis generalised) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 4. LAURA<SUP>a</SUP> 시험에서 보고된 이상반응
LAURA시험에서, 시험 치료기간의 중앙값은 이 약 군의 환자에서 24.0개월, 위약군의 환자에서 8.3개월이었다.
a 무작위 배정된 치료로서 1회 용량 이상 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란, 각막내피결손, 각막염, 점상각막염 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질성 폐렴 포함.
e 1건의 CTCAE 5등급(치명적)이 보고됨.
f 입 궤양, 구내염 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성 발진(rash vesicular), 피부미란(skin erosion) 포함.
h 손발톱바닥장애(nail bed disorder), 손발톱바닥감염(nail bed infection), 손발톱바닥염증(nail bed inflammation), 손발톱변색(nail discolouration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부 건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k QTcF 연장이 >500 msec를 보인 환자의 발생률을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
m LAURA 시험의 베이스라인 혈액 크레아티닌 청소율(<30 mL/min)은 이 약의 다른 단독요법 시험들에서의 베이스라인 혈액 크레아틴 청소율(<50 mL/min)보다 낮으므로, 등급 변화가 일어날 가능성이 더 높았다.
표 5. FLAURA<SUP>a</SUP> 시험에서 보고된 이상반응
FLAURA 시험에서 시험 치료기간의 중앙값은 이 약 군의 환자에서 16.2 개월이었고 EGFR TKI 대조약 군의 환자에서 11.5개월이었다.
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0.
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
e EGFR TKI 대조약 군에서 1건의 CTCAE 5등급(치명적) 사건이 보고되었다.
f 입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
i손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis), 전신 소양증(pruritis generalised) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
l실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 6. FLAURA2<SUP>a</SUP> 시험에서 보고된 이상반응
FLAURA2 시험에서 시험 치료기간의 중앙값은 이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 군의 환자에서 22.3 개월이었고 이 약 단일요법 군의 환자에서 19.3개월이었다.
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질화 폐렴(organising pnuemonia) 포함.
e 1건의 CTCAE 5등급(치명적) 사건이 보고되었다.
f 입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular) 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
I 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 7. AURA3<SUP>a</SUP>시험에서 보고된 이상반응
AURA3 시험에서 시험 치료기간의 중앙값은 이 약 군의 환자에서 8.1개월이었고 항암화학요법 군의 환자에서 4.2개월이었다.
a 최소 1회 투여를 한 환자의 사례만 요약되어 있다.
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
e CTCAE 5등급(치명적) 1건이 보고되었다.
f입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 농포성 발진(rash pustular) 포함.
h 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 건조증(xerosis) 포함.
i손발톱바닥장애(nail bed disorders), 손발톱바닥염증(nail bed inflammation), 손발톱바닥압통(nail bed tenderness), 손발톱변색(nail discoloration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱능성형성(nail ridging), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱주위염(paronychia) 포함.
j 눈꺼풀 소양증, 소양증, 전신 소양증 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
단일군 임상2상 AURAex 및 AURA2 임상시험에서의 안전성 결과는 일반적으로 AURA3의 이 약 군에서 관찰된 것과 일치하였다. 추가적이거나 예상하지 못한 독성이 관찰되지 않았으며, 이상반응은 종류, 중증도 및 빈도 등이 알려진 것과 유사하였다.
선택된 이상반응에 대한 설명
혈액학적 독성
이 약으로 치료를 받은 환자에서 실험실적 백혈구, 림프구, 호중구 및 혈소판 수의 중앙값에 조기 감소가 관찰되었으며, 이는 시간이 지남에 따라 안정해진 후 정상 하한보다 높게 유지되었다. 백혈구 감소증, 림프구 감소증, 호중구 감소증, 및 혈소판 감소증 이상사례가 보고되었으며, 대부분은 경증 또는 중등증이었고 투여 중지로 이어지지 않았다.
3) 국내 시판 후 수집된 중대한 이상사례 분석▪평가 결과 확인된 이상사례는 다음과 같다. 다만, 이로써 곧 해당성분과 다음의 이상사례 간에 인과관계가 입증된 것을 의미하는 것은 아니다.
◦ 호흡기계 : 폐색전증
※ 국내 시판 후 조사 결과
국내에서 1178명을 대상으로 실시한 시판 후 조사 결과, 이상사례의 발현율은 인과관계와 상관없이 69.10%(814/1178명, 2287건)로 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응 및 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 발현 빈도에 따라 아래 표에 나열하였다.
국내 시판 후 이상사례(재심사 이상사례 포함) 보고자료(1989‑2025.03.31.)를 토대로 실마리정보 분석▪평가 결과 추가적으로 확인된 이상사례는 다음과 같다. 다만, 이로써 곧 해당성분과 다음의 이상사례 간에 인과관계가 입증된 것을 의미하는 것은 아니다.
각종 신경계 장애 ‑ 미각 이상
근골격 및 결합 조직 장애 ‑ 등허리 통증, 사지 통증, 근육 연축
대사 및 영양 장애 ‑ 식욕 감소
전신 장애 및 투여 부위 병태 ‑ 무력증, 피로
4. 일반적 주의
1) EGFR 변이 상태 및 검사
비소세포폐암 환자에서 완전 종양 절제술 후 보조 치료로서 이 약의 사용을 고려할 때, EGFR 변이 양성 상태 (엑손 19 결손 또는 엑손 21[L858R] 치환 변이)는 치료 적격성을 나타낸다.
이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
절제 불가능한 국소 진행성(III기) 비소세포폐암 환자에서 이 약의 사용을 고려할 때, EGFR 돌연변이 양성 상태(엑손 19 결손 또는 엑손 21 [L858R] 치환 변이)는 치료 적격성을 나타낸다. 이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
국소 진행성 또는 전이성 비소세포폐암의 치료제로서 이 약의 사용을 고려할 때, 이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
단독요법에서 보조 치료 임상시험에서는 EGFR 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였다.
단독요법 및 페메트렉시드와 백금 기반 항암화학요법과 병용 요법의 1차 치료 임상시험에서는 EGFR 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였고, 인증받은 지역 실험실 검사도 사용되었다.
임상시험에서는 T790M 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였다.
2) 이 약은 운전 및 기계 조작하는 능력에 영향을 미치지 않거나 또는 현저한 영향을 미치지 않는다.
5. 상호작용
1) 이 약의 혈장 농도를 증가시킬 수 있는 활성 물질
생체 외(in vitro) 시험에서 이 약의 1단계 대사는 주로 CYP3A4 및 CYP3A5를 통해 일어나는 것으로 확인되었다. 약동학적 임상시험에서, 이 약과 이트라코나졸(강력한 CYP3A4 저해제) 1일 2회 200mg를 병용투여 하였을 때 이 약의 노출(커브아래면적(AUC;Area Under the Curve)은 24%(90% CI 15, 35) 증가하였고 C<SUB>max</SUB>는 ‑20%(90% CI ‑27, ‑13) 감소)에는 임상적으로 유의한 영향을 미치지 않았다. 따라서 CYP3A4 저해제는 이 약의 노출량에 영향을 미치지 않는 것으로 보인다.
2) 이 약의 혈장 농도를 감소시킬 수 있는 활성 물질
강력한 CYP3A4 유도제는 오시머티닙의 노출을 감소시킬 수 있다. 환자를 대상으로 한 약동학적 임상시험에서, 리팜피신(21일간 600mg매일 투여)과 병용투여 시 이 약의 정상상태의 AUC는 ‑78%(90% CI ‑81, ‑76) 감소하였다. 이 약과 강력한 CYP3A 유도제(예, 페니토인, 리팜피신, 카바마제핀, 세인트존스워트(St. John’s Wort))와의 병용투여는 피하는 것이 권장된다.
3) 위산을 감소시키는 활성 성분이 이 약의 대사 미치는 영향
환자를 대상으로 한 약동학적 임상시험에서, 오메프라졸과의 병용투여는 이 약의 노출량에 대하여 임상적으로 관련 있는 변화를 야기하지 않았다. 위내 pH를 변화시키는 약물들은 제한 없이 이 약과 병용투여 할 수 있다.
4) 이 약에 의해 혈장 농도가 변할 수 있는 활성 성분
생체 외 시험에 근거하였을 때, 이 약은 BCRP 수송체의 경쟁적 저해제이다. 이 약은 breast cancer resistant protein(BCRP) 및 P‑gp 기질의 노출을 증가시킬 수 있다.
환자를 대상으로 한 약동학적 임상시험에서, 이 약과 로수바스타틴(민감한 BCRP 기질)의 병용투여는 로수바스타틴의 AUC와 C<SUB>max</SUB>를 각각 35%(90% CI 15, 57), 72%(90% CI 46, 103) 증가시켰다.
BCRP에 약물 분포가 의존적이며 치료영역이 좁은 약물을 병용 투여하는 환자는 초기용량 설정 시 약동학적 상호작용 가능성을 면밀히 고려하고 이 약 투여 중 병용약물의 노출량 증가에 기인하여 발생할 수 있는 내약성 변화의 징후에 대해 주의 깊게 모니터링 해야 한다.
환자를 대상으로 한 약동학적 임상시험에서, 이 약과 심바스타틴(민감한 CYP3A4 기질)의 병용투여는 심바스타틴의 AUC와 C<SUB>max</SUB>를 각각 ‑9% (90% CI ‑23, 8), ‑23 % (90% CI ‑37, ‑6) 감소시켰다. 이 변화는 작고, 임상적으로 유의하지 않은 것으로 보인다. CYP3A4 기질과의 임상적 약동학적 상호작용은 없는 것으로 보인다.
임상 약동학 시험에서, 이 약과와 펙소페나딘(PXR/P‑gp 기질)의 병용 투여는 펙소페나딘의 AUC 및 C<SUB>max</SUB>를 단회 투여 후 각각 56% (90% CI 35, 79) 및 76% (90% CI 49, 108) 증가시켰고 항정상태에서 각각 27% (90% CI 11, 46) 및 25% (90% CI 6, 48) 증가시켰다. P‑gp에 약물 분포가 의존적이며 치료 영역이 좁은 약물(예, 디곡신, 다비가트란, 알리스키렌)을 병용 투여하는 환자는 이 약 투여 중 병용 약물의 노출 증가에 기인한 내약성 변화의 징후에 대해 주의 깊게 모니터링 되어야 한다.
6. 임부 및 수유부에 대한 투여
1) 남성 및 여성에서의 피임
가임 여성은 이 약 투여 중 임신을 피하여야 한다. 환자는 이 약 투여 완료 후 여성은 최소 2개월, 남성은 최소 4개월 동안 효과적인 피임법을 계속 사용하도록 해야 한다.
2) 임부
이 약을 임부에게 투여한 자료는 없거나 제한적이다. 동물시험에서는 생식독성을 보였다.
작용기전 및 비임상시험 데이터에 근거할 때, 이 약은 임신한 여성에게 투여 시 태아 독성을 유발할 수 있다. 임신한 랫드에게 오시머티닙 투여 시, 사람에게 예상되는 노출량과 유사한 수준에서 배자 치사, 태자 성장 감소 및 신생자(neonate) 사망과 관련이 있는 것으로 나타났다. 이 약은 임부 및 피임을 하지 않고 임신할 가능성이 있는 여성에게 투여하지 않는다.
3) 수유부
오시머티닙 또는 그 대사체가 사람 모유로 분비되는지는 알려져 있지 않다.
임신한 랫드 및 초기 수유 중인 랫드에의 이 약의 투여는 성장율 감소와 신생자 사망(neonatal death)을 포함한 이상반응과 관련이 있었다. 이 약 또는 그 대사체가 동물의 모유로 분비되는지에 대한 정보는 충분하지 않다. 젖먹이 아이에게의 위험을 배제할 수 없다. 수유부에게 이 약 치료 중 수유를 중단하도록 해야 한다.
4) 수태능
사람의 수태능에 이 약이 미치는 영향에 대한 자료는 없다. 동물실험 결과 이 약이 남성 및 여성 생식기관에 영향을 주어 생식능력을 저하시킬 수 있는 것으로 나타났다.
7. 소아에 대한 투여
소아 환자에서 이 약의 안전성 ◦유효성은 확립되지 않았다.
8. 고령자에 대한 투여
집단 약동학 분석에서 연령은 오시머티닙의 노출량에 영향을 주지 않는 것으로 나타났다.
ADAURA, FLAURA, FLAURA2 및 AURA (이 약의 단독요법 군 (N = 1813))에서, 42 % 환자가 65세 이상이었고, 11%가 75세 이상이었다. 젊은 환자(65세 미만)와 비교하여, 65세 이상의 환자에서 시험약 용량 변경이 필요한 이상반응이 더 많이 보고되었다(일시중단 또는 용량감소)(14% vs 10%). 보고된 이상반응의 종류는 연령에 관계없이 유사하였다. 고령의 환자가 젊은 환자에 비해 3등급 이상의 이상반응을 더 많이 보고하였다(11% vs 9%). 고령의 환자와 젊은 환자 간 유효성에 전반적인 차이는 관찰되지 않았다.
9. 간장애 환자에 대한 투여
임상시험에 근거할 때 경증(Child Pugh A) 또는 중등증 간장애(Child Pugh B) 환자에서의 용량 조절은 필요하지 않다. 이와 유사하게, 집단 약동학 분석에 근거할 때, 경증의 간장애 환자(총빌리루빈≦정상 상한치[ULN] 이고 AST >ULN 또는 총빌리루빈이 1.0배의 ULN과 1.5배의 ULN 및 모든 수치의 AST 사이) 또는 중등증의 간장애 환자(총빌리루빈이 1.5배의 ULN과 3배의 ULN 및 모든 수치의 AST 사이)에서 용량 조절은 필요하지 않다.
중증의 간장애 환자에서 이 약의 안전성 ◦유효성은 확립되지 않았다.
10. 신장애 환자에 대한 투여
임상시험 및 집단 약동학 분석에 근거할 때, 경증, 중등증 또는 중증의 신장애 환자에게 용량조절은 필요하지 않다.
말기 신질환 환자 [Cockcroft and Gault 방정식에 의해 계산된 크레아티닌 청소율 (CLcr)이 15 mL/min 미만] 또는 투석 중인 환자에 대한 이 약의 안전성 ◦유효성은 확립되지 않았다. 중증 및 말기 신장애 환자 치료 시 주의해야 한다.
11. 과량투여시의 처치
이 약을 사용한 임상시험에서 제한된 수의 환자들이 이 약 1일 최대 240mg까지 용량 제한적 독성(dose limiting toxicities)없이 투여되었다. 이 연구들에서, 이 약을 1일 160mg 및 240mg 투여받은 환자들은 80mg을 투여받은 환자들보다 전형적인 EGFR‑유도 이상반응(주로 설사와 피부발진)의 빈도 및 중증도의 증가를 경험하였다.
이 약의 과량투여 시 특별한 치료방법이 없다. 의사는 일반적인 보조 조치를 실시하고 대증적으로 치료해야 한다.
12. 보관 및 취급상의 주의사항
1) 어린이의 손이 닿지 않는 곳에 보관한다.
2) 다른 용기에 바꾸어 넣는 것은 사고원인이 되거나 품질 유지 면에서 바람직하지 않으므로 이를 주의한다.
13. 전문가를 위한 정보
1) 약리작용
이 약은 티로신 키나제 억제제 (Tyrosine Kinase Inhibitor, TKI)이다. 이 약은 감작 돌연변이 (EGFRm) 및 TKI‑내성 돌연변이 T790M을 가진 표피 성장인자 수용체 (EGFRs)의 강력한 선택적 비가역적 경구 억제제이다.
In vitro 시험에서 이 약은 임상적으로 관련된 모든 EGFR 감작 돌연변이체 및 T790M 돌연변이체 비소세포폐암 (NSCLC) 다양한 세포주에 대하여 높은 효력 및 억제 활성을 나타내는 것을 입증하였다. (phospho‑EGFR에 대한 겉보기 IC<SUB>50</SUB>6 nM에서 54 nM) 이로 인해 세포 성장이 억제되는 반면, 야생형 세포주에서는 EGFR에 대한 활성이 유의하게 낮게 나타난다. (phospho‑EGFR에 대한 겉보기 IC<SUB>50</SUB>480 nM에서 1.9 μM) In vivo에서 이 약의 경구 투여는 EGFRm 및 T790M 모두의 NSCLC 이종이식 및 유전자 이식 마우스 폐 종양 모델에서 종양 축소를 나타난다.
심장 전기생리학
이 약의 QTc 간격 연장 가능성은 AURA2에서 오시머티닙 1일 80 mg을 투여 받은 210 명의 환자에서 평가되었다. QTc 간격에 대한 오시머티닙의 영향을 평가하기 위해 연속적 ECG를 단회 투여 및 정상상태에서 수집했다. 이 약의 약동학/약력학 분석은 80 mg에서 14 msec의 약물 관련 QTc 간격 연장을 상한 16 msec (90% CI)으로 예측했다.
2) 약동학적 정보
오시머티닙 약동학 파라미터는 건강한 시험대상자 및 NSCLC 환자들에서 확인되었다. 집단 약동학 분석에 근거하여, 오시머티닙 겉보기 혈장 청소율은 14.3 L/h이며, 겉보기 분포 용적은 918 L이고, 말단 반감기는 약 44시간이다. 이 약과 페메트렉시드 및 백금 기반 항암화학요법을 병용하여 치료한 환자의 약동학은 이 약 단독요법으로 치료받은 환자의 약물동태와 유사하다. AUC 및 Cmax 는 20에서 240mg 용량 범위에서 용량에 비례하여 증가하였다. 이 약을 1일 1회 투여한 결과, 약 3배의 축적을 나타내며, 정상 상태 노출은 투여 15일에 도달되었다. 정상 상태에서, 순환혈장농도는 일반적으로 24시간 투여 간격 동안 1.6배 범위 내에서 유지된다.
흡수
이 약을 경구 투여한 후, 오시머티닙의 최고 혈장 농도는 t<SUB>max</SUB>중앙값(최소‑최대)은 6시간 (3‑24)에 도달되었으며, 일부 환자들에서 처음 24시간 동안 여러 개의 피크가 관찰되었다. 이 약의 절대 생체 이용률은 70%이다. (90% CI 67, 73) 80 mg을 투여 받은 환자에 대한 임상 약동학 시험에 근거하여, 음식은 오시머티닙의 생체 이용률을 임상적으로 의미 있는 정도로 변경하지 않는다. (AUC 6% 증가 (90% CI ‑5, 19) 및 C<SUB>max</SUB>‑7%감소 (90% CI ‑19, 6)). 5일간 오메프라졸 투여에 의해 위장 pH가 상승한 건강한 시험대상자에서 80 mg 정제를 투여했을 때, 오시머티닙 노출은 영향을 받지 않았으며 (AUC 및 C<SUB>max</SUB>증가 각각 7% 및 2%), 노출비에 대한 90% CI는 80‑125% 한도 내에 포함되었다.
분포
모집단에서 평가된 오시머티닙의 정상 상태에서의 평균 분포 용적은 (V<SUB>ss</SUB>/F)918L이며, 이는 조직으로의 광범위한 분포를 시사한다. In vitro에서, 오시머티닙의 혈장 단백질 결합은 94.7%이다. (비결합 5.3%) 오시머티닙은 또한 랫드와 사람의 혈장 단백질, 사람 혈청 알부민 및 랫드와 사람 간세포에 공유 결합하는 것으로 확인되었다.
생체 내 변환
In vitro 시험에서 오시머티닙은 주로 CYP3A4 및 CYP3A5에 의해 대사되는 것을 보여준다. In vitro 시험에 근거하여, 두가지 약리학적 활성 대사체 (AZ7550 및 AZ5104)가 이 약을 경구 투여한 후 전임상 종의 혈장 및 사람에서 이후 확인되었다; AZ7550은 이 약과 유사한 약리학적 프로파일을 나타낸 반면, AZ5104는 돌연변이 및 야생형 EGFR 모두에서 더 큰 효력을 보였다. 환자들에게 이 약을 투여한 후 두 대사체 모두 혈장에서 서서히 나타났으며, t<SUB>max</SUB>중앙값 (최소‑최대)은 각각 24 (4‑72) 및 24 (6‑72) 시간이었다. 사람 혈장에서, 모성분 오시머티닙은 0.8%를 차지하였고, 두 대사체는 총 방사능의 0.08% 및 0.07%에 기여하였으며, 방사능의 대부분은 혈장 단백질에 공유 결합하였다. AUC에 근거한 AZ5104 및 AZ7550의 기하 평균 노출은 정상 상태에서 오시머티닙 노출의 각각 약 10%였다.
오시머티닙의 주요 대사 경로는 산화 및 탈알킬화였다. 사람의 뇨 및 분변 통합 검체에서 최소 12개의 성분들이 관찰되었으며, 5개 성분은 투여량의 >1%를 차지하였고, 이 중 미변화 오시머티닙, AZ5104, 및 AZ7550은 투여량의 약 1.9, 6.6, 및 2.7%를 차지한 반면, cysteinyl 부가생성물 (M21), 및 알려지지 않은 대사체 (M25)는 투여량의 각각 1.5% 및 1.9%를 차지하였다.
In vitro 시험에 근거하여 오시머티닙은 임상적으로 관련된 농도에서 CYP 3A4/5의 경쟁적 억제제이지만, CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 및 2E1 에 대해서는 아니다. In vitro 시험에 근거하여 오시머티닙은 간에서 임상적으로 관련된 농도에서 UGT1A1 및 UGT2B7의 억제제가 아니다. UGT1A1의 장에서의 억제는 가능하지만, 임상적 영향은 알려지지 않았다.
배설
20 mg 단회 경구 투여 후, 투여량의 67.8%가 분변으로 회수된 반면 (모성분으로 1.2%), 투여량의 14.2% (모성분으로 0.8%)는 84일의 표본 수집 시까지 소변에서 발견되었다. 미변화된 오시머티닙은 배설의 약 2%를 차지하였으며, 0.8%는 소변으로, 1.2%는 분변으로 배설되었다.
특수 집단
집단 기반 PK 분석에서 (n=1367), 예측된 정상 상태 노출 (AUC<SUB>ss</SUB>)과 환자의 연령 (범위: 25‑91세), 성별 (65% 여성), 인종 (백인, 아시아인, 일본인, 중국인 및 비아시아‑비백인 환자), 치료 차수 및 흡연 상태 (n=34 현재 흡연자, n=419 과거 흡연자) 간에 임상적으로 유의한 관련성은 확인되지 않았다. 집단 PK 분석에서 체중이 유의한 공변량으로 나타났으며, 체중 중앙값 61 kg의 AUC<SUB>ss</SUB>와 비교했을 때, 88 kg에서 43 kg 체중 범위에서 오시머티닙 AUC<SUB>ss</SUB>변화가 20% 미만으로 예상된다. (95%‑5% 사분위) 체중의 극단을 고려하여 43 kg 미만에서 88 kg 초과까지 AZ5104 대사체 비는 11.8% 에서 9.6% 범위이며, AZ7550의 경우 그 범위는 12.8%에서 8.1%이다. 집단 PK 분석에 근거하여 혈청 알부민은 유의한 공변량으로 확인되었으며, 베이스라인 알부민 중앙값 39 g/L에 대한 AUC<SUB>ss</SUB>와 비교했을 때 알부민 범위 29에서 46 g/L의 알부민 범위에서 오시머티닙 AUCss 변화는 30% 미만으로 예상된다. (95% ‑ 5% 사분위수) 체중 또는 베이스라인 알부민 차이에 의한 이러한 노출 변화는 임상적으로 관련이 없는 것으로 고려된다.
뇌 전이 환자
뇌 전이가 있는 EGFR 돌연변이 양성 NSCLC 환자 (n=4)를 대상으로 한 마이크로도즈 PET 연구에서 오시머티닙의 뇌 침투 및 분포(중앙값 T<SUB>max</SUB>: 22분;평균C<SUB>max</SUB>: 투여된 용량의 1.5%가 뇌에 도달)는 건강한 지원자 연구에서 관찰된 것과 유사했다 (n=7; 중앙값 T<SUB>max</SUB>: 11분; 평균 C<SUB>max</SUB>: 투여된 용량의 2.2%가 뇌에 도달).
3) 임상적 유효성 및 안전성
이전 보조 화학 요법을 받았거나 받지 않은 EGFR 변이 양성 비소세포폐암의 보조 치료 ‑ ADAURA
이전 보조 화학 요법을 받았거나 받지 않은 완전 종양 절제술을 받은 EGFR 변이 양성 (엑손 19 결손 또는 L858R 치환 변이) 비소세포폐암 환자의 보조 치료에 대한 이 약의 유효성 및 안전성은 무작위 이중 맹검 위약 대조 시험 (ADAURA)에서 입증되었다.
절제 가능한 종양 (IA기 제외)이 있는 환자들은 중앙 실험실에서 생검 또는 수술 검체를 사용하여 전향적으로 실시된 cobas EGFR Mutation Test에 의해 확인된 EGFR 엑손 19 결손 또는 엑손 21 L858R 치환 변이가 있어야 했다.
환자들은 무작위 배정되어 (1:1) 이 약 80 mg 경구 1일 1 회 또는 위약을 수술에서 회복된 후 투여 받고 제공된 경우 표준 보조 화학 요법을 받았다. 보조 화학 요법을 받지 않은 환자들은 수술 후 10 주 이내에, 보조 화학 요법을 받은 환자들은 수술 후 26 주 이내에 무작위 배정되었다. 무작위 배정은 돌연변이 유형 (엑손 19 결손 또는 엑손 21 L858R 치환 변이), 민족성 (아시아인 또는 비아시아인) 및 AJCC 7판에 따른 pTNM (IB 또는 II 또는 IIIA) 기반 병기 별로 계층화되었다. 질병이 재발하거나 수용할 수 없는 독성이 나타날 때까지 또는 3년 동안 투여를 받았다.
주요 유효성 결과 측정은 연구자 평가에 따른 무병 생존 기간 (disease‑free survival, DFS)이었다. 추가적 유효성 결과 측정은 DFS 비율, 전체 생존기간 (overall survival, OS), OS 비율 및 건강 관련 삶의 질 (HRQL) SF‑36이 악화되는 시간을 포함한다.
총 682 명의 환자가 이 약 (n = 339) 또는 위약 (n = 343)에 무작위 배정되었다. 중앙 연령은 63세 (30‑86세 범위)였고 11%는 75세 이상; 70%는 여성, 64%는 아시아인, 72%는 흡연 미경험자였다. 베이스라인 WHO 수행 능력은 0 (64%) 또는 1 (36%)이었다; 31%는 IB기, 34% II기, 그리고 35%는 IIIA기였다. EGFR 돌연변이 상태와 관련하여, 55%는 엑손 19 결손이었고 45%는 엑손 21 L858R 치환 변이였다; 9명의 환자 (1%)는 de novo T790M 변이도 있었다. 환자의 대다수 (60%)는 무작위 배정 전 보조 화학 요법을 받았다 (26% IB; 71% IIA; 73% IIB; 80% IIIA).
II‑IIIA기 집단과 전체 집단 (IB‑IIIA)에 대해 DFS 분석을 했다. ADAURA는 위약으로 치료받은 환자 대비 이 약으로 치료받은 환자에 대해 질병 재발 또는 사망의 위험이 통계적으로 유의하고 임상적으로 의미있는 감소함을 입증하였다. 이 약으로 치료받은 II‑IIIA기 질환 환자들은 위약 대비 질병 재발 또는 사망의 위험이 83% 감소했다 (중앙값은 각각 계산되지 않음 (not calculated, NC) 및 19.6개월, HR=0.17, 99.06% CI:0.11, 0.26; P<0.0001). 위약 대비 이 약으로 치료 받은 전체 집단 (IB‑IIIA)은 질병 재발 또는 사망의 위험이 80% 감소한 것으로 나타났다 (중앙값은 각각 NC 및 27.5개월, HR=0.20, 99.12% CI:0.14, 0.30; P<0.0001).
이 약에서 질병이 재발한 환자는 37명이었다. 가장 흔하게 보고된 재발 부위는 다음과 같다; 폐 (19 명); 림프절 (10명) 및 CNS (5명). 위약으로 질병이 재발한 환자는 157명이었다. 가장 흔하게 보고된 부위는 다음과 같다; 폐 (61명); 림프절 (48명) 및 CNS (34명).
OS의 최종 분석은 II‑IIIA기 집단 (21.3% 성숙; HR=0.49; 95.03% CI: 0.33, 0.73; p‑값=0.0004) 및 전체 집단 (IB‑IIIA; 18.2% 성숙; HR=0.49; 95.03% CI: 0.34, 0.70; p 값 < 0.0001) 둘 모두에 대해 위약과 비교하여 이 약을 투여받은 환자의 51% 사망 위험 감소를 보이며 임상적으로 의미 있고 통계적으로 크게 유의한 OS 개선을 입증하였다. 전체 집단 (IB‑IIIA)에서, 중도절단된 환자의 추적관찰 시간의 중앙값은 두 치료군 모두 61.5개월이었다.
연구자 평가에 따른 ADAURA의 유효성 결과는 표 8 및 9에 요약되어 있으며, II‑IIIA기 환자 및 전체 집단 (IB‑IIIA)의 DFS 및 OS 에 대한 Kaplan‑Meier 곡선은 그림 1부터 그림 4에 제시하였다.
표 8. 연구자 평가에 따른 II‑IIIA기 환자에서의 유효성 결과
HR (Hazard Ratio)=위험비; CI (Confidence Interval)=신뢰 구간; NC (Not Calculable)=계산 불가능
DFS 결과는 연구자 평가에 근거한 것이다.
HR <1은 이 약에 유리하다.
DFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 22.1개월이었고 위약을 투여 받은 환자에서 14.9개월이었다.
중도절단된 환자의 OS에 대한 추적관찰 시간의 중앙값은 이 약 투여 군(stage II‑IIIA 집단)에서 61.7개월, 위약 군에서 60.4개월이었다.
DFS 결과는 1차 분석(2020년 1월 17일)에서 나왔다. OS 결과는 최종 분석(2023년 1월 27일)에서 나왔다.
a 중간분석 (33% 성숙)을 위해 조정된 p‑값 <0.0094이 통계적 유의성을 달성하기 위해 필요했다.
b 36개월 시점에 추적 관찰 환자 (patient at risk) 수는 이 약 투여군은 18명이고, 위약 군은 9명이었다.
c 중간 분석을 위해 조정된 통계적 유의성을 달성하려면 p‑값 <0.0497이 필요했다.
d Kaplan‑Meier 방법으로 계산했다.
그림 1. 연구자 평가에 따른 무병 생존 기간 (II‑IIIA기 환자)의 Kaplan‑Meier 곡선
HR (Hazard Ratio)=위험비; CI (Confidence Interval)=신뢰 구간; NC (Not Calculable)=계산 불가능
DFS 결과는 연구자 평가에 근거한 것이다.
HR <1은 이 약에 유리하다.
DFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 22.1개월이었고 위약을 투여 받은 환자에서 16.6개월이었다.
중도절단된 환자의 OS에 대한 추적관찰 시간의 중앙값은 이 약 투여 군 및 위약 군에서 모두 61.5개월이었다.
DFS 결과는 1차 분석(2020년 1월 17일)에서 나왔다. OS 결과는 최종 분석(2023년 1월 27일)에서 나왔다.
a 중간분석 (29% 성숙)을 위해 조정된 p‑값 < 0.0088이 통계적 유의성을 달성하기 위해 필요했다.
b 36개월 시점에 추적 관찰 환자 (patient at risk) 수는 이 약 투여 군은 27명이고, 위약 군은 20명이었다.
c 중간 분석을 위해 조정된 통계적 유의성을 달성하려면 p‑값 < 0.0497이 필요했다.
d Kaplan‑Meier 방법으로 계산했다.
그림 3. 연구자 평가에 따른 무병 생존 기간 (전체 집단)의 Kaplan‑Meier 곡선
위약 환자 대비 이 약 환자에 대한 CNS DFS (CNS 재발 또는 사망까지의 시간)의 탐색적 분석에서 임상적으로 의미있는 개선이 전체 집단에서 HR 0.18 (95% CI:0.10, 0.33; P <0.0001)로 관찰되었으며, 이는 위약 대비 이 약 군에서 CNS 질환 재발 또는 사망의 위험이 82% 감소함을 나타낸다.
환자 보고 결과
ADAURA에서 건강 관련 삶의 질 (HRQL)은 Short Form (36) Health Survey version 2 (SF‑36v2) 설문지를 사용하여 평가되었다. SF‑36v2는 치료가 완료되거나 중단될 시까지 무작위 배정과 관련하여 12주, 24주, 그 후 24주마다 관리되었다. 전반적으로, HRQL은 두 군 모두에서 유지되었으며, II‑IIIA기 집단에서 75%를 넘는 환자가 SF‑36의 물리적 구성요소에서 임상적으로 의미있는 악화 또는 사망을 경험하지 않았거나 (이 약 대 위약에 대해 75.1% 대 83.5%) 또는 SF‑36의 정신적 구성요소에서 임상적으로 의미있는 악화 또는 사망 (이 약 대 위약에 대해 77.7% 대 78.1%)을 경험하지 않았다. 이 약 군에서 SF‑36의 물리적 구성요소 또는 사망에 대한 약화 시간 (time to deterioration, TTD)이 짧은 경향이 관찰되었으며 (HR=1.43, 95% CI:0.96, 2.13), 두 군 모두에서 중앙값 TTD에 도달하지 않았다. SF‑36의 정신적 구성요소 또는 사망에 대한 TTD에서 군 간의 차이는 없었으며 (HR=0.90, 95% CI: 0.61, 1.33), 중앙값 TTD는 이 약 군에서 39.0개월 (95% CI: NC, NC)이고 위약 군에서는 도달하지 않았다.
절제 불가능한 국소 진행성(III기) EGFR 돌연변이 양성 비소세포폐암 – LAURA
확정적 백금‑기반 화학 방사선 요법 중 또는 후 진행되지 않은, EGFR 돌연변이 양성의 절제 불가능한 국소 진행성(III기) 비소세포폐암 환자의 치료에 대한 이 약의 유효성 및 안전성은 무작위 배정, 이중 눈가림, 위약 대조 시험(LAURA)에서 입증되었다. 환자들은 동시적 화학 방사선 치료(CCRT)나 순차적 화학 방사선 치료(SCRT) 요법을 받았으며, 여기에서 최소 2주기 또는 주 5 회 용량의 백금‑기반 화학 요법과 총 60 Gy ±10% (54 Gy ‑ 66 Gy) 용량의 방사선 치료가 무작위 배정 전 6주 이내에 완료되었다. 환자 종양 조직 검체는 공인/승인된 시험을 이용하여 중앙 또는 현지 검사로 확인된 EGFR 엑손 19 결손 또는 엑손 21 L858R 변이가 있어야 했다. 확정적 백금‑기반 화학 방사선 요법 중 또는 후 2등급 이상의 폐염증이 있거나 또는 화학 방사선 요법 이전에 간질성 폐염증의 병력이 있는 환자는 제외되었다.
환자들은 이 약 80 mg을 1일 1회 경구로(n=143) 또는 위약(n=73)을 투여받도록 무작위 배정되었다(2:1). 무작위 배정은 선행 화학 방사선 치료 전략(CCRT vs SCRT), 화학 방사선 치료 전 종양 병기 결정(IIIA vs IIIB/IIIC), 및 중국 코호트에 따라 층화되었다. 환자들은 치료에 대한 불내성 또는 확인된 질병 진행까지 시험 치료를 계속 받아야 했다. 이 후, 모든 환자는 치료 의사의 소견에 따라 기대되는 임상적 유익성이 있는 경우 현지 임상진료에 따라 라벨이 공개된 이 약을 제공받았다.
일차 유효성 평가변수는 눈가림된 독립적 중앙 검토(BICR)로 평가된 PFS였다. 추가 유효성 평가변수에는 CNS PFS, OS, ORR, DoR, 사망 또는 원격 전이까지 걸린 시간(TTDM), 첫 번째 후속 치료 시작 후 두 번째 PFS (PFS2), 첫 번째 후속 치료 또는 사망까지 걸린 시간(TFST), 및 두 번째 후속 치료 또는 사망까지 걸린 시간(TSST)이 포함되었다. CNS PFS, ORR, DoR, 및 TTDM은 모두 BICR로 평가되었다. 또한 환자 보고 결과도 평가되었다.
전체 시험대상군의 베이스라인 인구학적 및 질병 특성은 다음과 같았다: 연령 중앙값 63세(범위 36‑84세), ≧75세(13%), 여성(61%), 아시아인(82%), 백인(14%), 흡연력이 없는 사람(70%). 베이스라인 WHO 수행 능력 상태는 0 (51%) 또는 1 (49%)이었다; 환자의 35%는 IIIA기, 49%는 IIIB기, 그리고 16%는 IIIC기 비소세포폐암이었다. 환자의 조직학적 특성으로는 96%는 비편평세포암이었고, 4%는 편평세포암이었다. EGFR 돌연변이 상태와 관련해서는 54%가 엑손 19 결손이었고 45%가 엑손 21 L858R 변이였다. 무작위 배정 전에 환자의 89%는 CCRT를 받았고 11%는 SCRT를 받았다. 모든 환자는 백금‑기반 화학 요법(55%는 카보플라틴‑기반 화학 요법, 44%는 시스플라틴‑기반 화학 요법)을 받았다. 총 방사선량의 중앙값은 두 군에서 모두 환자에서 60 Gy였다.
백금‑기반 화학 방사선 요법 후 이 약의 치료는 위약 대비 PFS의 통계적으로 유의하고 임상적으로 유의미한 개선을 가져왔다(56% 성숙; HR=0.16; 95% CI: 0.10, 0.24; P<0.001, 각각 중앙값 39.1개월 및 5.6개월). 6, 12, 18, 24 및 36개월에서 이 약으로 치료 받은 환자의 무진행 생존 비율(각각 84%, 74%, 67%, 65% 및 58%)은 위약으로 치료받은 환자(각각 45%, 22%, 14%, 13% 및 10%)의 무진행 생존 비율보다 더 컸다.
임상시험 계획서에 따라, 모든 환자는 베이스라인 자기 공명 영상(MRI) 뇌 스캔을 받았고, 치료 중에 1명을 제외하고 모두 예정된 MRI 뇌 스캔을 받았다. 위약과 비교해 이 약으로 치료받은 환자에서 CNS PFS (신경방사선학자 BICR 평가에 근거)의 명목상 통계적으로 유의하고 임상적으로 유의미한 개선이 있었다(27% 성숙; HR=0.17; 95% CI: 0.09, 0.32; P<0.001 [명목]). 위약군에 비해 이 약 군에서 더 낮은 비율의 환자가 신경방사선학자 검토에 따라 새로운 CNS 병변이 있었다(각각 17/143 [12%] vs 26/73 [36%]).
전체 생존 기간의 중간 분석에서, 이 약에 유리한 긍정적 경향이 있었지만(20% 성숙; HR=0.81; 95% CI: 0.42, 1.56; P=0.530) 통계적으로 유의하지 않았다.
LAURA 임상시험에서 얻은 유효성 결과는 표 10에 요약되어 있으며, PFS와 CNS PFS에 대한 Kaplan‑Meier 곡선은 각각 그림 5 및 6에 나타내었다.
표 10. LAURA에서 얻은 유효성 결과
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, CNS PFS, ORR, DoR, 및 TTDM 결과는 BICR에 따라 평가하였다.
모든 환자에서 PFS에 대한 추적관찰 시간 중앙값은 이 약 군에서 22.0개월, 위약군에서 5.6개월이었다.
HR <1 및 오즈비 >1은 이 약에 유리하다.
a 중간 분석(20% 성숙)에 대해 보정한 결과 통계적 유의성에 도달하기 위해서는 p 값<0.00036이 필요했다.
b 확인되지 않은 응답을 기반으로 했다.
c 분석은 화학 요법 전 병기(IIIA vs IIIB/IIIC)에 따라 계층화된 로지스틱 회귀 분석을 사용하여 수행되었다.
d Kaplan‑Meier 방법으로 계산했다.
그림 5. LAURA에서 BICR로 평가된 PFS에 대한 Kaplan‑Meier 곡선
위약 대비 이 약의 PFS 유익성은 성별, 무작위 배정 시점의 연령, 흡연력, 민족성, 선행 화학 방사선 치료 전략(CCRT vs SCRT), 화학 방사선 치료 전 병기(IIIA vs IIIB/IIIC), 선행 화학 방사선 치료에 대한 반응, 및 EGFR 돌연변이 유형(엑손 19 결손 또는 L858R)을 포함하여 분석된 사전 정의된 모든 하위군에서 일관적이었다.
그림 6. LAURA에서 BICR로 평가된 CNS PFS의 Kaplan‑Meier 곡선
이 약에 무작위 배정된 환자는 위약에 무작위 배정된 환자에 비해 PFS2, TFST 및 TSST에서 임상적으로 유의미한 개선이 있었다. 이러한 진행 후 평가변수의 분석은 이 약으로 교차 투여 수준이 높음에도 불구하고 PFS 유익성이 후속 차수 치료를 통해 유지됨을 입증하였다.
환자 보고 결과
환자 보고 증상 및 HRQL 자료는 EORTC QLQ‑C30 (C30) 및 폐암 모듈 EORTC QLQ‑LC13 (LC13)을 이용하여 전자적으로 수집되었다. 베이스라인에서 환자 보고 증상, 신체적 기능 및 전반적인 건강 상태/삶의 질(GHS/QoL)은 이 약 군과 위약군 간에 유사하였다. 환자가 보고한 5가지 주요 폐암 및 치료 관련 증상(기침, 호흡 곤란, 흉통, 피로, 및 식욕 상실)과 신체적 기능 영역 및 GHS/QoL의 베이스라인 대비 전체 평균 변화에서 임상적으로 유의미한 차이는 없었다. 치료군 간에 이러한 환자 보고 증상, 신체적 기능 또는 GHS/QoL의 악화 위험에서 유의미한 차이는 관찰되지 않았다.
이전에 치료 받은 적 없는 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 – FLAURA –단독요법
진행성 질환에 대하여 이전에 전신 치료를 받지 않은 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 치료 시 이 약의 유효성 및 안전성이 무작위배정, 이중 눈가림, 활성 대조 시험(FLAURA)에서 입증되었다.
환자들은 이 약 (n=279, 80 mg 1일 1회 경구 투여) 또는 EGFR TKI 대조약(n=277; 게피티닙 250 mg 1일 1회 경구 투여 또는 엘로티닙 150 mg 1일 1회 경구 투여)을 투여 받도록 1:1로 무작위 배정 되었다. 무작위 배정은 EGFR 변이 유형(Ex19del 또는 L858R) 및 민족성(아시아인 또는 비아시아인)에 따라 층화되었다. 환자들은 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더 이상 임상적 유익성을 경험하지 않을 때까지 시험 치료를 투여 받았다. EGFR TKI 대조약을 투여 받은 환자의 경우, 진행이 발생한 후 종양 샘플 검사 시 T790M 변이에 양성인 경우 이 약 공개라벨으로 교차가 허용되었다.
이 약은 EGFR TKI 대조약과 비교하여 임상적으로 의미 있고 통계적으로 크게 유의한 PFS 개선을 입증하였다 (중앙값 각각 18.9 개월 및 10.2 개월, HR=0.46, 95% CI: 0.37, 0.57; P<0.0001). 연구자 평가에 의한 FLAURA에서의 유효성 결과가 표 11에 요약되어 있으며, PFS에 대한 Kaplan‑Meier 곡선을 그림 7에 나타내었다. 전체 생존 기간 최종 분석 (58% 성숙)은 EGFR TKI 대조약과 비교하여 이 약에 무작위 배정된 환자에서 0.799의 HR (95% CI: 0.641, 0.996; P = 0.0462)로 통계적으로 유의한 개선 및 임상적으로 의미 있는 보다 긴 중앙 생존 기간을 입증했다 (표 11 및 그림 8). 12개월, 18개월, 24개월 및 36개월 시점에 생존한 환자 비율은 이 약 (각각 89%, 81%, 74% 및 54%) 치료 시 EGFR TKI 대조약 (각각 83%, 71%, 59% 및 44%) 치료에 비해 높았다.
표 11. 연구자 평가에 따른 FLAURA에서의 유효성 결과
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, ORR, DoR 결과는 RECIST 연구자 평가에 근거한 것이다.
*확인되지 않은 반응에 근거한 것이다.
PFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 15.0개월이었고 EGFR TKI 대조약을 투여 받은 환자에서 9.7개월이었다.
OS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 35.8개월이었고 EGFR TKI 대조약을 투여 받은 환자에서 27.0개월이었다.
PFS, ORR, DoR 결과는 자료마감일 (data cut‑off) 2017년 6월 12일이다. OS 결과는 자료마감일 2019년 6월 25일이다.
HR <1 및 오즈비 >1은 이 약에 유리하다.
a 중간 분석(25% 성숙)을 위해 조정된 통계적 유의성을 달성하려면 p‑값 <0.0495이 필요했다
b 눈가림된 독립적 중앙 검토(BICR)에서 평가한 ORR 결과는 연구자 평가에서 보고된 것과 일치하였다; BICR 평가에 따른 ORR은 이 약에서 78% (95% CI: 73, 83)였고 EGFR TKI 대조약에서 70% (95% CI: 65, 76)였다.
그림 7. FLAURA에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
FLAURA2 – 병용요법
진행성 질환에 대하여 이전에 전신 치료를 받지 않은 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 치료 시 페메트렉시드와 백금 기반 항암화학요법과 병용하는 이 약의 유효성 및 안전성이 무작위배정, 공개, 활성 대조 시험 (FLAURA2)에서 입증되었다. 환자의 종양 조직 샘플은 현지 또는 중앙 검사로 확인된, EGFR TKI 민감도와 관련이 있는 것으로 알려진 두 가지 일반적인 EGFR 변이 (Ex19del 또는 L858R) 중 하나가 있어야 했다.
환자들은 다음의 치료군 중 하나에 무작위 배정되었다 (1:1):
‧ 이 약 (80 mg) 1일 1회 경구 투여 시, 페메트렉시드 (500 mg/m2) 그리고 시스플라틴 (75 mg/m2) 또는 카보플라틴 (AUC5) 중 연구자가 선택한 약물을 4주기 동안 21일 주기의 Day1에 정맥 내 투여한 후, 이 약 (80 mg) 1일 1회 경구 투여 및 페메트렉시드 (500 mg/m2) 3주마다 정맥 내 투여 (n=279)
‧ 이 약 (80 mg) 1일 1회 경구 투여 (n=278)
무작위 배정은 인종 (중국인/아시아인, 비중국인/아시아인 또는 비아시아인), WHO 수행 능력 (0 또는 1), 및 조직 검사 방법 (중앙 또는 현지)에 따라 층화되었다. 환자는 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더 이상 임상적 유익성을 경험하지 않을 때까지 시험 치료를 투여 받았다.
일차 유효성 평가변수는 RECIST 1.1에 따라 연구자가 평가한 PFS였다. 추가적인 유효성 평가변수에는 연구자의 평가에 따른 OS, ORR, DoR, PFS2, TFST 및 TSST가 포함되었다. BICR에 의해 평가된 CNS PFS, CNS ORR, CNS DoR 그리고 PRO도 평가되었다.
전체 시험 집단에 대한 베이스라인 인구통계학적 특성 및 질병 특성은 다음과 같았다: 연령 중앙값 61세 (범위 26‑85세), ≧75세 (8%), 여성 (61%), 아시아인 (64%), 백인 (28%), 흡연 미경험자 (66%). 모든 환자는 WHO 수행 능력이 0 또는 1이었다; 환자의 49%는 전이성 골 질환이 있었고, 환자의 53%는 흉곽 외 전이가 있었으며 20%는 간 전이가 있었다. 환자의 41%는 CNS 전이가 있었다 (베이스라인에 CNS 병변 부위, CNS 전이에 대한 병력, 및/또는 이전 수술, 및/또는 이전 방사선요법을 근거로 연구자가 확인함).
페메트렉시드 및 백금 기반 항암화학요법과 병용하는 이 약은 이 약의 단일요법과 비교하여 임상적으로 의미 있고 통계적으로 유의한 PFS 개선을 입증하였다 (51% 성숙; HR=0.62, 95% CI: 0.49, 0.79; P<0.0001; 중앙값 각각 25.5개월 및 16.7개월). OS 중간 분석 시점에, 페메트렉시드 및 백금 기반 항암화학요법과 병용한 이 약 투여 군의 환자들은 이 약의 단일요법 군에 비해 OS에 영향을 받지 않았다 (27% 성숙; HR=0.90, 95% CI: 0.65, 1.24; P = 0.5238).
연구자 평가에 따른 FLAURA2의 유효성 결과는 표 12에 요약되어 있으며, PFS에 대한 Kaplan‑Meier 곡선은 그림 9에 나타내었다.
표 12. 연구자 평가에 따른 FLAURA2에서의 유효성 결과
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, ORR, DoR, DCR 결과는 RECIST 연구자 평가에 근거한 것이다.
PFS 추적관찰 시간의 중앙값은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 받은 환자에서 19.5개월이었고 이 약의 단독요법 군에서는 16.5개월이었다.
HR <1 및 오즈비 >1은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에 유리하다.
a 중간 분석(27% 성숙)에 따르면 통계적 유의성을 달성하려면 p‑값 < 0.00158 이 필요했다.
b 확인되지 않은 응답을 기반으로 했다.
c BICR에서 평가한 ORR 결과는 연구자 평가에서 보고된 것과 일치하였다; BICR 평가에 따른 ORR은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에서 92% (95% CI: 88, 95)였고 이 약의 단독요법 군에서 83% (95% CI: 78, 87)였다.
d 분석은 인종 (중국인/아시아인 vs. 비중국인/아시아인 vs. 비아시아인), WHO 수행 능력 (0 또는 1), 및 조직 검사 방법 (중앙 또는 현지)에 따라 계층화된 로지스틱 회귀 분석을 사용하여 수행되었다.
e Kaplan‑Meier 방법으로 계산했다.
그림 9. FLAURA2에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
이 약의 단일요법 대비 페메트렉시드 및 백금 기반 항암화학요법과 병용하는 이 약의 PFS 유익성은 민족성, 연령, 성별, 흡연 유무, 시험 시작 시 CNS 전이 상태, 및 EGFR 돌연변이 유형 (엑손 19 결손 또는 L858R)을 포함하여 사전에 정의된 모든 분석 하위군에 걸쳐 일관성 있게 관찰되었다.
베이스라인 대비 표적 병변 크기의 가장 우수한 백분율 변화 중앙값은 페메트렉시드 및 백금 기반 항암화학요법과 이 약 병용 투여 군에서 ‑53% (범위: 100%~20%)였고 이 약의 단일요법 군에서는 ‑50% (범위: ‑100%~40%)였다.
1차 치료로서 페메트렉시드 및 백금 기반 항암화학요법과 이 약 병용 투여 군에 무작위 배정된 환자도 이 약의 단일요법 군에 무작위 배정된 환자에 비해 임상적으로 의미있는 PFS2, TFST 및 TSST 개선을 보였다. 이러한 진행 후 평가변수 분석은 PFS 유익성이 후속 차수의 치료까지 대부분 유지되었음을 입증하였다.
FLAURA2에서 CNS 전이 유효성 데이터
스테로이드가 필요하지 않은 무증상 CNS 전이 환자와 최종 치료 및 스테로이드 치료 완료 후 최소 2주 동안 신경학적 상태가 안정적인 환자는 FLAURA2 연구에 무작위 배정될 자격이 있었다. 모든 환자는 베이스라인 뇌 스캔이 가능했다. 수정된 RECIST를 사용한 BICR 평가에서는 CNS 측정 가능 및/또는 측정 불가능 병변(cFAS)이 있는 환자 222/557명(40%)의 하위 그룹과 CNS 측정 가능 병변(cEFR)이 있는 환자 78/557 (14%) 의 하위 그룹이 추가로 생성되었다. FLAURA2에 대한 BICR의 CNS 효능 평가 관련하여, 이 약의 단독요법 군에 비해 페메트렉시드와 백금 기반 항암화학요법과 이 약의 병용 투여 군에서 cFAS 및 cEFR에 대한 CNS PFS에서 임상적으로 의미 있는 개선을 입증했다(cFAS: 27% 성숙도, HR= 0.58, 95% CI 0.33, 1.01, P=0.0548[명목] 및 cEFR: 37% 성숙도, HR=0.40, 95% CI 0.19, 0.84, P=0.0157[명목]).
CNS 효능 데이터는 표 13에 요약되었다.
표 13. FLAURA2에서 베이스라인 뇌 스캔에서 CNS 전이가 있는 환자에 대해 BICR 평가된 CNS 유효성
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
HR <1 및 오즈비 >1은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에 유리하다.
a 확인되지 않은 응답을 기반으로 했다.
b 명목(Nominal) P‑값
c 분석은 치료 요인을에 대한 로지스틱 회귀분석을 이용하여 수행했다.
베이스라인 대비 표적 CNS 병변 크기의 최고 백분율 변화 중앙값은 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서 ‑94%(범위: ‑100% ~ 7%)였고, 이 약의 단독요법 군에서 ‑61%(범위: ‑100% ~ 68%)이었다.
FLAURA2에서는 CNS 전이 상태(기준시 CNS 병변 부위, 병력 및/또는 이전 수술 및/또는 CNS 전이에 대한 이전 방사선 치료를 기반으로 연구자가 식별함)를 기반으로 사전 지정된 PFS 하위군으로 연구를 시작했다. 연구 시작 시 CNS 전이 상태와 관계없이, 이 약의 단독요법 군에 비해 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서의 PFS 개선이 입증되었다. 연구 시작 시 CNS 전이 상태별 PFS에 대한 Kaplan‑Meier 곡선을 그림 10에 제시했다.
그림 10. FLAURA2에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
환자 보고 증상 및 HRQL은 EORTC QLQ C30 및 그에 대한 폐암 모듈 (EORTC QLQ‑LC13)을 이용해 전자적으로 수집되었다. LC13은 첫 8주 동안 주1회 시행되었고, 이후 진행 전에는 3주마다 그리고 진행 후에는 8주마다 시행되었다. C30은 첫 8주 동안 3주마다 평가되었고, 이후 진행 전에는 6주마다 그리고 진행 후에는 8주마다 평가되었다. 베이스라인에, 환자 보고 증상, 신체 기능 또는 GHS/QoL에서 페메트렉시드 및 백금기반 화학요법과 병용하는 이 약 군과 이 약의 단일요법군 간에 차이는 관찰되지 않았다. 첫 19개월에 걸친 순응도는 전반적으로 높았고 (≧80%) 두 군 모두 유사했다.
주요 폐암 증상 분석
베이스라인부터 제19개월까지 수집된 자료는 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군과 이 약의 단일요법 군에서 사전 명시된 5가지 일차 PRO 증상 중 3가지 (기침, 호흡곤란, 및 흉통)에 대해 유사한 개선을 나타냈으며, 기침에서의 개선은 두 군 모두 확립된 임상적으로 관련된 기준치 (베이스라인 대비 변화 ≦‑10)에 도달했다. 이 약의 단일요법군에서는 식욕 상실 및 피로에 대해 개선되는 추세를 보였다. 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서는 화학요법의 첫 4주기 동안 피로가 악화되는 추세를 보이다가 이후 개선되는 추세를 보였으며, 식욕 상실은 악화되는 추세를 보였다. 이러한 변화는 임상적으로 의미가 없었다. 이러한 자료는 표 14에 제시되어 있다.
표 14. 혼합 모델 반복 측정 ‑ 주요 폐암 증상 – 이 약 단독 요법으로 치료받은 환자와 페메트렉시드 및 백금 기반 항암화학요법과 이 약을 병용하여 치료받은 환자의 베이스라인으로부터의 평균 변화
전반적인 건강상태/QoL 및 신체기능 개선 분석
두 군 모두 신체 기능 및 GHS/QoL에 임상적으로 의미 있는 변화가 없다고 보고했다.
이전에 치료 받은 적 있는 T790M양성 비소세포폐암 환자 ‑ AURA3
TKI 치료 후 또는 치료 중 진행되면서 국소진행 또는 전이된 T790M 비소세포폐암 환자 치료 시 이 약의 유효성 및 안전성은 무작위 배정된, 공개, 활성 대조군 임상 3상시험(AURA 3)에서 증명되었다.
환자들은 이 약 (n=279) 또는 백금기반 이중 항암화학요법제(n=140)에 2:1 (이 약: 백금기반 이중 항암화학요법제) 비율로 무작위 배정되었다. 무작위 배정은 민족성 (아시아인 및 비아시아인)에 따라 층화되었다. 이 약 군의 환자는 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더이상 임상적 유익성을 경험하지 않을 때까지 이 약 80 mg을 1일 1회 경구 투여 받았다.
화학요법은 최대 6주기동안 매 21일 주기의 제 1일째에 페메트렉시드 500mg/m2와 카보플라틴 AUC5 병용요법 또는 페메트렉시드 500mg/m2와 시스플라틴 75mg/m2병용요법으로 이루어졌다. 4주기의 백금기반 화학요법 후에 질병이 진행되지 않은 환자는 페메트렉시드 유지요법을 받을 수 있었다 (매 21일 주기의 제 1일째에 페메트렉시드 500mg/m2). 화학요법군의 시험대상자 중 객관적인 방사선학적 진행이 있는 환자 (연구자에 의해 그리고 독립적인 중앙 영상 검토에 의해 확정된)는 이 약으로 치료를 시작할 수 있는 기회가 주어졌다.
AURA 3시험에서 항암화학요법군 대비 이 약으로 치료받은 환자군에서 통계적으로 유의한 PFS 개선이 증명되었다. AURA 3 연구자 평가에 따른 AURA 3의 유효성 결과는 표 15에 요약되어 있고, 그림 11에 PFS에 대한 Kaplan‑Meier 곡선을 나타내었다. 최종 OS 분석 (67 % 성숙 시 실시)에서 치료군 간에 통계적으로 유의한 차이가 관찰되지 않았으며, 이 시점에는 항암화학요법에 무작위 배정된 99명의 환자 (71 %)가 이 약 치료로 넘어가 있었다.
표 15. 연구자 평가에 따른 AURA3에서의 유효성 결과
HR=위험비; CI=신뢰구간; OS=전체 생존기간
RECIST 연구자 평가에 근거한 모든 유효성 결과
HR <1 및 오즈비 >1은 이 약에 유리하다.
1 OS의 최종 분석은 67% 성숙 시 실시되었다. HR에 대한 CI는 이전 중간 분석을 위해 조정되었다. OS 분석은 교차에 대한 잠재적인 교란혼재효과는 보정되지 않았다(항암화학요법군의 99명 [71%] 환자는 이후 오시머티닙 치료를 받았다.).
2 연구자 평가에 의한 ORR 및 DoR 결과는 눈가림된 독립적 중앙 검토(Blinded Independent Central Review, BICR)를 통해 보고된 것과 일치한다; BICR 평가에 의한 ORR은 오시머티닙 치료 시 64.9% [95% CI: 59.0, 70.5], 항암화학요법제 치료 시 34.3% [95% CI: 26.5, 42.8]였다. ; BICR에 의한 DoR은 오시머티닙 치료 시 11.2개월(95% CI: 8.3, NC), 항암화학요법제 치료 시 3.1 개월(95% CI: 2.9, 4.3)이었다.
그림 11. AURA3에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
항암화학요법제를 투여 받은 환자군과 비교하여 이 약을 투여 받은 환자군에게 유리한 0.50 미만 HR을 보이는 임상적으로 의미 있는 PFS 개선은 민족성, 나이, 성별, 흡연 유무, 시험 시작 시 CNS 전이 상태, EGFR 변이 (엑손 19 결손 또는 L858R), 및 EGFR‑TKI 1차 치료 기간을 포함하여 사전에 정의된 모든 분석 하위군에서 일관성 있게 관찰되었다.
4) 독성시험 정보
반복 투여 독성
랫드와 개에 대한 반복 투여 독성시험에서 관찰된 주요 결과는 눈(각막), 위장관(혀 포함), 피부 및 수컷과 암컷 생식기관의 상피에 영향을 미치는 위축성, 염증성 그리고/또는 퇴행성 변화로 구성되었다. 이러한 결과는 80 mg 치료 용량의 환자들에서 관찰된 것보다 낮은 혈장 농도에서 발생하였다. 투여 1개월 후 발생한 결과는 투여 중단 후 1개월 이내에 대체로 회복을 나타내었다.
발암성 및 돌연변이성
오시머티닙은 26주 동안 Tg rasH2 유전자 이식 마우스에게 경구 투여했을 때 발암 가능성을 보이지 않았다.
랫드의 104주 발암성 연구에서 1일 1회 80 mg의 권장 임상 용량에서 관찰된 AUC의 0.2배 노출에서 수정체 섬유 변성 및 장간막 림프절에서 증식성 혈관 병변 (혈관종 과형성 및 혈관종)의 발생률 증가가 관찰되었다. 수정체 섬유 변성은 52주차에 처음 관찰되고 투여 기간이 증가함에 따라 발생률 및 중증도가 점진적으로 증가하는 수정체 혼탁에 대한 검안경 관찰과 일치했다. 이러한 결과의 임상적 연관성은 배제할 수 없다. 장간막 림프절에서의 증식성 혈관 병변(혈관종 과형성 및 혈관종)은 인간의 혈관 신생물에 대한 발암 가능성을 예측하는 것은 아니다.
오시머티닙은 in vitro 및 in vivo 시험에서 유전적 손상을 유발하지 않았다.
생식 독성
동물에 대한 시험에 근거하여 수컷 수태능은 이 약 치료로 손상될 수 있다. 1개월 이상 오시머티닙에 노출된 랫드와 개에서 퇴행성 변화를 고환에서 보였으며, 3개월 동안 오시머티닙에 노출된 후 랫드에서 수컷 수태능이 감소하였다. 이러한 결과들은 임상적으로 관련된 혈장 농도에서 관찰되었다. 1개월 투여 후 보인 고환에서의 병리학적 소견은 랫드에서 가역적이었으나, 개에서 이러한 병변의 가역성에 대한 확정적인 진술은 할 수 없다.
동물에서의 시험에 근거하여 암컷 수태능은 이 약 치료로 손상될 수 있다. 반복투여 독성시험에서 임상적으로 관련된 혈장 농도로 오시머티닙에 1개월 이상 노출된 랫드에서 발정휴지기 증가, 난소에서 황체 퇴행 및 자궁과 질의 상피 얇아짐이 관찰되었다. 1개월 투여 후 관찰된 난소에서의 결과는 가역적이었다. 랫드에 대한 암컷 수태능 시험에서, 오시머티닙 20 mg/kg/day 투여 (권장 1일 임상 용량인 80 mg과 거의 동등한 용량)는 발정 주기 또는 임신하는 암컷의 수에 영향이 없었으나, 초기 배아 사망을 야기했다. 이러한 결과는 1개월간 휴약 후 가역성의 증거를 보였다.
랫드에 대한 수정된 배태자 발달 시험에서, 오시머티닙은 임신한 랫드에게 배아 착상 전 투여 시 배아 치사를 초래했다. 이러한 영향은 권장 용량 1일 80 mg에서의 사람 노출과 동등한 노출 (총 AUC에 근거)인 20 mg/kg/day의 모체 내약 용량 (maternally tolerated dose)에서 관찰되었다. 기관 형성기에 20 mg/kg 이상의 용량에 대한 노출은 태자 무게 감소를 유발하였으나, 태자의 외형 또는 내장 형태에는 유해한 영향을 미치지 않았다. 오시머티닙을 임신한 암컷 랫드에게 임신 기간에 걸쳐서, 그리고 이후 수유 초기에 투여했을 때, 수유를 받는 차산자에서 오시머티닙 및 그 대사체에 대한 노출을 보였으며, 차산자의 생존 감소 및 차산자 성장 감소가 있었다. (20 mg/kg 이상의 용량에서)
1) 간질성 폐질환(Interstitial Lung Disease, ILD)
ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들에서 이 약을 단독요법으로 투여받은 1813명의 환자 중 간질성 폐질환 또는 간질성 폐질환 유사 이상반응(예. 폐염증)은 4.0%의 환자에게서 보고되었으며, 이 중 치명적인 건은 0.4%(n=7) 보고되었다. 간질성 폐질환은 일본인 환자의 11.2%, 일본인이 아닌 아시아인 환자의 2.3%, 비아시아인 환자의 2.7%에게서 발생하였다. 첫 번째 투여부터 간질성 폐질환 또는 간질성 폐질환 유사 이상반응들의 발현시점의 중앙값은 2.8개월이었다.
FLAURA2에서 페메트렉시드 및 백금 기반 항암화학요법과 병용하여 이 약을 투여받은 276명의 환자 중 간질성 폐질환 또는 간질성 폐질환 유사 이상반응은 3.3%에서 보고되었으며, 이 중 치명적인 건은 0.4% (n=1)이었다. 간질성 폐질환은 일본인 환자의 14.9% 및 비아시아인 환자의 1.7%에게서 발생하였다. FLAURA2 병용요법 군에서 일본인이 아닌 아시아인 환자에서는 간질성 폐질환이 발생하지 않았다. 첫 번째 투여부터 간질성 폐질환 또는 간질성 폐질환 유사 이상반응들의 발현시점의 중앙값은 5.3개월이었다.
간질성 폐질환을 시사할 수 있는 호흡기 증상(예: 호흡곤란, 기침, 발열)의 악화가 나타나는 환자는 이 약을 중단하고 즉시 간질성 폐질환에 대해 조사한다. 간질성 폐질환이 확인되면 이 약을 영구 중단한다.
확정적 백금‑기반 화학 방사선 요법 후 간질성 폐질환
LAURA 임상시험에서 확정적 백금‑기반 화학 방사선 요법 후, 이 약을 투여받은 143명의 환자 중 56%(80명)와 위약을 투여받은 73명의 환자 중 38%(28명)에서 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응(예: 폐염증)이 보고되었다. 이 약 군에서는 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응 중 0.7%(1명)의 치명적 증례가 보고되었으며, 3등급은 3.5%, 2등급은 34%, 1등급은 18%이었다. 이 약을 투여받은 환자 중 7%가 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응으로 투여를 영구 중단 했으며, 35%는 일시 중단했다. 재투여를 받은 환자 46명 중 11%는 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 재발하였다. 이 약을 투여받고 방사선 폐렴을 포함한 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생한 80명의 환자 중 40%는 회복, 1.3%는 후유증을 동반하고 회복, 16%는 회복 중이었으며, 41%는 회복되지 않고 1.3%(1명)는 사망하였다.
확정적 백금‑기반 화학 방사선 요법 후 이 약으로 치료받은 환자에서 간질성 폐질환을 시사할 수 있는 호흡기 증상(예: 호흡곤란, 기침, 발열)의 악화가 나타나는 환자는 이 약을 중단하고 즉시 간질성 폐질환에 대해 조사한다. 간질성 폐질환이 확인되면 무증상(1등급) 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생하는 경우, 적절히 이 약을 지속하거나 일시 중지 및 재개한다. 2등급 이상의 간질성 폐질환 또는 간질성 폐질환 유사 이상반응이 발생하는 경우, 이 약의 사용을 영구 중단한다.
2) 스티븐스‑존슨증후군, 다형성 홍반 (Erythema multiforme) 및 독성 표피 괴사 용해
이 약 치료와 관련하여, 다형성 홍반 및 독성 표피 괴사 용해의 사례 보고는 흔하지 않게 보고되었고, 스티븐스‑존슨증후군은 드물게 보고되었다. 치료를 시작하기 전에 환자에게 스티븐스‑존슨증후군, 다형성 홍반 및 독성 표피 괴사 용해의 징후와 증상을 알려야 한다.
스티븐스‑존슨증후군 또는 독성 표피 괴사 용해를 연상시키는 징후와 증상이 나타나면, 이 약을 일시 중단해야 한다. 스티븐스‑존슨증후군 또는 독성 표피 괴사 용해가 진단되면, 이 약을 즉시 중지해야 한다.
다형성 홍반을 연상시키는 징후와 증상이 있는 경우, 긴밀한 환자 모니터링 및 이 약의 중단 또는 중지를 고려해야 한다.
3) QTc 간격 연장
이 약 단독요법 (80mg)으로 치료한 ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들의 1813명 환자 중, 1.1%(n=20)는 500 msec 초과 QTc를, 4.3%(n=78)는 베이스라인 QTc로부터 60 msec를 초과하는 QTc 증가를 나타냈다. 이 약에 대한 약동학적 분석 시 QTc 간격 연장의 농도 의존적 증가가 예측되었다. ADAURA, LAURA, FLAURA, FLAURA2 또는 AURA 임상시험들에서 QTc와 관련된 부정맥 사례는 보고되지 않았다.
가능한 경우, 선천적으로 긴 QT 증후군이 있는 환자는 이 약의 사용을 피한다 (3.이상반응 참조). 울혈성 심부전, 전해질 이상이 있는 환자, 또는 QTc 간격이 연장되는 것으로 알려진 약물을 투여 중인 환자에서는 심전도(ECG) 및 전해질의 주기적인 모니터링을 고려한다. 심전도 검사에서 QTc 간격이 500 msec을 초과하는 결과가 적어도 2회 별도로 발생한 환자에서는 QTc 간격이 481msec 미만이 될 때까지, 또는 베이스라인 QTc가 481msec 이상인 경우 베이스라인으로 회복될 때까지 이 약을 중단하고, 이후에 용법용량 항 표 1에 기술된 바와 같이 감소된 용량으로 투여를 재개한다. 다음 중 어느 하나와 더불어 QTc 간격 연장이 발생한 환자는 이 약을 영구 중단한다: 염전성 심실빈맥(Torsade de pointes), 다형성 심실빈맥(polymorphic ventricular tachycardia), 중대한 부정맥의 징후/증상.
4) 심수축성/심장독성 변화
임상시험들에서 이 약을 투여받은 1813명의 환자 중 3.8%에서 심장독성(심부전, 만성 심부전, 울혈성 심부전, 폐부종, 박출률 감소)을 나타냈다. 이 중 1명(0.1%)은 치명적인 결과로 이어졌다.
임상시험에서 베이스라인 및 최소 1회의 좌심실박출률 추적 평가를 받은 이 약의 단독요법으로 치료받은 환자의 4.2%(65/1557)에서 좌심실 박출량이 10퍼센트포인트 이상 감소하고 50% 미만으로 나타났다. 위약 대조 시험(ADAURA)에서 이 약을 투여 받은 환자의 1.5%(5/325)와 위약을 투여 받은 환자의 1.5%(5/331)에서 10%포인트 이상 50% 미만의 좌심실박출률 감소를 경험했다. LAURA 임상시험에서 백금‑기반 화학 방사선 요법 후, 이 약으로 치료받은 환자의 3.0% (4/135명)와 위약으로 치료받은 환자 0명에서 좌심실 박출량이 10퍼센트포인트 이상 감소하고 50% 미만으로 나타났다. FLAURA2에서 베이스라인 및 최소 1회의 좌심실박출률 추적 평가를 받은 이 약과 페메트렉시드, 백금 기반 항암화학요법을 병용하여 치료받은 환자의 8.0%(21/262)에서 좌심실 박출량이 10%포인트 이상 감소하고 50% 미만으로 나타났다.
심장 위험인자가 있거나 좌심실박출률에 영향을 미칠 수 있는 상태의 환자에게는 베이스라인 및 치료 중 좌심실박출률 평가를 포함하는 심장 모니터링을 한다. 치료 중 관련된 심장 징후/증상이 있는 환자의 경우, 좌심실박출률 평가를 포함한 심장 모니터링을 한다. 좌심실박출률이 기저치 대비 10 퍼센트포인트 이상 감소하고 50% 미만으로 나타날 경우, 이 약 투여를 최대 4주간 중단하고 중단 후에도 증상이 개선되지 않을 때에는 이 약 투여를 영구 중단한다. 증상성 울혈성 심부전 발생 시에는 이 약의 투여를 영구 중단한다.
5) 각막염
ADAURA, FLAURA, FLAURA2 및 AURA 임상시험들에서 이 약의 단독요법으로 치료된 1813명의 환자 중 0.6%(n=10)에서 각막염이 보고되었다. 다음과 같은 각막염의 징후 및 증상을 보이는 환자들은 즉시 안과전문의에게 상담을 받아야 한다: 눈의 염증, 눈물 분비, 빛에 민감한 눈, 시야 흐림, 눈의 통증 그리고/또는 눈의 충혈 (용법용량 항 참조)
6) 재생 불량성 빈혈
재생 불량성 빈혈이 이 약의 치료와 관련하여 드물게 보고되었다. 일부 사례는 치명적인 결과가 있었다. 치료를 시작하기 전에 환자에게 지속적인 발열, 멍, 출혈, 창백을 포함하되 이에 국한되지 않는 재생 불량성 빈혈의 징후와 증상에 대해 알려야 한다. 재생 불량성 빈혈을 암시하는 징후 및 증상이 발생하면 환자를 면밀히 모니터링하고 이 약의 중지 또는 중단을 고려해야 한다. 재생 불량성 빈혈이 확인된 환자는 이 약을 중단해야 한다.
2. 다음 환자에는 투여하지 말 것.
이 약의 주성분이나 부형제에 대해 중증의 과민증이 있는 환자
3. 이상반응
1) 안전성 프로파일의 전반적인 요약
EGFR 변이 양성 비소세포폐암 환자에 대한 시험
이 약 단독요법의 안전성은 EGFR 변이 양성 비소세포폐암 환자 1813명으로부터 수집된 데이터를 기반으로 한다. 이러한 환자들은 4건의 무작위배정된 3상 임상시험(ADAURA: 보조 치료; FLAURA 및 FLAURA2(단독요법 군):1차 치료; AURA3: 2차 치료), 2건의 단일군 2상 임상시험(AURAex 및 AURA2: 2차 치료 이상) 및 1건의 1상 임상시험(AURA1: 1차 치료 이상)에서 이 약 1일 80 mg을 투여 받았다.
대부분의 이상반응 중증도는 1등급 또는 2등급이었다. 가장 흔한 빈도로 보고된 약물이상반응은 설사(47%), 발진(46%), 손발톱주위염(34%), 건성 피부(32%), 및 구내염(24%)이었다. 이 약에서 나타난 3등급 및 4등급 이상반응은 각각 9.2%와 0.2%였다. 이 약 80 mg을 1일 1회 치료받은 환자들에게서, 약물이상반응으로 인한 용량감소는 3.6%의 환자들에게서 나타났다. 이상반응으로 인한 투약중단은 4.7%이었다.
백금‑기반 화학 방사선 요법 후 이 약(1일 1회 80 mg)의 안전성은 EGFR 돌연변이 양성 비소세포폐암 환자 143명에서 얻은 자료를 기반으로 한다.
페메트렉시드와 백금 기반 항암화학요법을 병용하여 투여한 이 약의 안전성은 EGFR 변이 양성 비소세포폐암 환자 276명 데이터를 기반으로 하며, 이 약의 단독요법 및 페멕트렉시드와 백금 기반 항암화학요법의 알려진 안전성 프로파일과 일치했다.
스테로이드 치료가 필요한 간질성 폐질환, 약물 유도 간질성 폐질환, 방사선 폐염증의 과거 병력이 있거나 임상적으로 활동성 간질성 폐질환 증거가 있는 환자는 임상시험에서 제외되었다. 안정 시 ECG (예를 들어, QTc 간격이 470 ms 초과)로 측정 시 리듬 및 전도가 임상적으로 중요한 비정상인 환자는 이러한 시험들에서 제외되었다. 스크리닝 및 이후 12주마다 환자의 좌심실박출률이 평가되었다.
AURAex 및 AURA2에 참여한 한국인 환자(66명)에서 전체 및 3/4등급 이상반응 발생율은 전체 환자군과 유사하였다. 다만, 한국인 환자에서 전체 환자군에 비해 상대적으로 많이 발생하는 이상반응은 발진, 소양증, 손발톱주위염, 혈소판 감소증, 백혈구 감소증으로 나타났다. 상대적으로 낮은 빈도로 발생하는 이상반응은 설사, 오심으로 보고되었다. 이들 이상반응은 대부분 1등급 또는 2등급이었다.
2) 이상반응 표
이상반응은 ADAURA, FLAURA, FLAURA2, AURA3, AURAex, AURA2, 및 AURA1 시험들에서 이 약의 단독요법으로 1일 80 mg을 투여 받은 EGFR 변이 양성 환자 1813명의 통합 데이터세트에서의 비교 가능한 이상반응 발생률에 근거하여 표 2에 빈도 카테고리로 분류되었다.
약물이상반응은 MedDRA 기관계 분류(System Organ Class;SOC)에 따라 나열하였다. 각 기관계 분류 내에서, 약물이상반응은 빈도 내림차순으로(즉, 가장 빈번한 반응이 제일 먼저 오도록) 나열하였다. 각 빈도 군 내에서, 약물이상반응은 중대성이 감소하는 순으로 나열하였다. 또한, 각 약물이상반응의 해당 빈도 범주는 CIOMS III 분류에 근거하여 다음과 같이 정의한다: 매우 흔하게(≧1/10), 흔하게(≧1/100 ~ <1/10), 흔하지 않게(≧1/1,000 ~ <1/100), 드물게(≧1/10,000 ~ <1/1000), 매우 드물게(<1/10,000), 빈도불명(사용 가능한 데이터로부터 추정할 수 없음).
표 2. ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 시험들에서 보고된 이상반응<SUP>a</SUP>
|
MedDRA SOC |
CIOMS 분류/전체 빈도(모든 CTCAE 등급b) |
CTCAE 3등급 이상의 빈도 |
|
MedDRA 선호 용어 | ||
|
혈액 및 림프계 이상 | ||
|
재생 불량성 빈혈 |
드물게(0.06%) |
0.06% |
|
눈 이상 | ||
|
각막염c |
흔하지 않게(0.6%) |
0.06% |
|
호흡계, 흉부 및 종격 이상 | ||
|
비출혈 |
흔하게(6%) |
0% |
|
간질성 폐질환d |
흔하게(4.0%)e |
1.4% |
|
위장관계 이상 | ||
|
설사 |
매우 흔하게(47%) |
1.4% |
|
구내염f |
매우 흔하게(24%) |
0.4% |
|
피부 및 피하조직 이상 | ||
|
발진g |
매우 흔하게 (46%) |
0.8% |
|
손발톱주위염h |
매우 흔하게 (34%) |
0.4% |
|
건성 피부i |
매우 흔하게 (32%) |
0.1% |
|
소양증j |
매우 흔하게 (17%) |
0.06% |
|
탈모증 |
흔하게(5%) |
0% |
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
흔하게(2.1%) |
0% |
|
두드러기 |
흔하게(1.9%) |
0.1% |
|
피부 과다 색소 침착k |
흔하게(1.0%) |
0% |
|
다형성 홍반l |
흔하지 않게(0.3%) |
0% |
|
독성 표피 괴사 용해m |
흔하지 않게(0.2%) | |
|
피부 혈관염m |
흔하지 않게(0.2%) | |
|
스티븐스-존슨증후군n |
드물게(0.02%) | |
|
실험실적 수치 | ||
|
혈액 크레아틴 인산 활성 효소 증가 |
흔하게(1.9%) |
0.3% |
|
QTc 간격 연장o |
흔하게(1.1%) | |
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||
|
백혈구 감소p |
매우 흔하게(65%) |
1.8% |
|
림프구 감소p |
매우 흔하게(64%) |
8% |
|
혈소판 수 감소p |
매우 흔하게(53%) |
1.3% |
|
호중구 감소p |
매우 흔하게(36%) |
4.0% |
|
혈액 크레아티닌 증가p |
흔하게(9%) |
0.2% |
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란, 각막내피결손, 각막염, 점상각막염 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질성 폐렴 포함.
e 7 건의 CTCAE 5등급(치명적)이 보고됨.
f 입 궤양 형성, 구내염 포함
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성 발진(rash vesicular), 피부미란(skin erosion) 포함.
h 손발톱바닥장애(nail bed disorder), 손발톱바닥감염(nail bed infection), 손발톱바닥염증(nail bed inflammation), 손발톱변색(nail discolouration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k 시판 후 조사에서 지속색소이상홍반 사례가 보고되었다.
l ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 시험의 1813명 환자 중 6명이 다형성 홍반을 보고하였다. 시판 후 조사 연구(N=3578)로부터의 7건을 포함하여, 다형성 홍반의 시판 후 보고도 있었다.
m 추정 빈도. 추정치에 대한 95% CI의 상한은 3/1813 (0.17%)이다. 임상시험에서 보고 없음.
n 시판 후 연구에서 보고된 한 건의 사례이며, 빈도는 ADAURA, FLAURA, FLAURA2(단독요법 군) 및 AURA 연구와 시판 후 연구(N=5391)에서 도출되었다.
o QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
p 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 3. ADAURA<SUP>a</SUP>시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=337) |
위약 (N=343) | ||||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%)c |
모든 등급 (%) |
3등급 이상 (%)c | ||
|
MedDRA 선호 용어 | ||||||
|
눈 이상 | ||||||
|
각막염d |
0.6 |
0 |
0.3 |
0 | ||
|
호흡계, 흉부 및 종격 이상 | ||||||
|
비출혈 |
5.6 |
0 |
0.9 |
0 | ||
|
간질성 폐질환e |
3.0 |
0 |
0 |
0 | ||
|
위장관계 이상 | ||||||
|
설사 |
46.3 |
2.4 |
19.8 |
0.3 | ||
|
구내염f |
28.2 |
1.8 |
6.4 |
0 | ||
|
피부 및 피하조직 이상 | ||||||
|
발진g |
39.2 |
0.3 |
19.0 |
0 | ||
|
손발톱주위염h |
36.5 |
0.9 |
3.8 |
0 | ||
|
건성 피부i |
29.4 |
0.3 |
7.3 |
0 | ||
|
소양증j |
19.3 |
0 |
8.7 |
0 | ||
|
탈모증 |
5.6 |
0 |
2.0 |
0 | ||
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
1.8 |
0 |
0 |
0 | ||
|
피부 과다 색소 침착 |
1.8 |
0 |
0 |
0 | ||
|
두드러기 |
1.5 |
0 |
0.3 |
0.3 | ||
|
실험실적 수치 | ||||||
|
혈액 크레아틴 인산 활성 효소 증가 |
3.3 |
0.9 | ||||
|
QTc 간격 연장k |
0.6 |
0 | ||||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||||||
|
백혈구 감소l |
54.0 |
0 |
25.4 |
0 | ||
|
혈소판 수 감소l |
47.2 |
0 |
6.6 |
0.3 | ||
|
림프구 감소l |
43.8 |
2.2 |
14.4 |
0.9 | ||
|
호중구 감소l |
25.6 |
0.3 |
10.2 |
0.3 | ||
|
혈액 크레아티닌 증가l |
9.8 |
0 |
4.5 |
0.3 | ||
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0
c 모든 사례는 3등급이었다. 사망은 없었다.
d 각막염(keratitis), 점상각막염(punctate keratitis), 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect) 포함.
e 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
f 구내염(stomatitis), 입 궤양 형성(mouth ulceration) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis), 전신 소양증(pruritis generalised) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 4. LAURA<SUP>a</SUP> 시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=143) |
위약 (N=73) | ||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
MedDRA 선호 용어 | ||||
|
눈 이상 | ||||
|
각막염c |
0.7 |
0 |
1.4 |
0 |
|
호흡계, 흉부 및 종격 이상 | ||||
|
간질성 폐질환d |
8 |
2.1e |
1.4 |
0 |
|
비출혈 |
0.7 |
0 |
0 |
0 |
|
위장관계 이상 | ||||
|
설사e |
36 |
2.1 |
14 |
0 |
|
구내염f |
15 |
0 |
4.1 |
0 |
|
피부 및 피하조직 이상 | ||||
|
발진g |
36 |
0.7 |
19 |
0 |
|
손발톱 주위염h |
23 |
0 |
1.4 |
0 |
|
건성 피부i |
17 |
0.7 |
5 |
0 |
|
소양증j |
13 |
0 |
7 |
0 |
|
탈모증 |
1.4 |
0 |
0 |
0 |
|
두드러기 |
1.4 |
0 |
1.4 |
0 |
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
0 |
0 |
0 |
0 |
|
피부 과다 색소 침착 |
0 |
0 |
0 |
0 |
|
다형성 홍반 |
0 |
0 |
0 |
0 |
|
실험실적 수치 | ||||
|
혈액 크레아틴 인산 활성 효소 증가 |
3.5 |
1.4 |
0 |
0 |
|
QTc 간격 연장k |
0.7 |
0 | ||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||||
|
림프구 감소l |
70 |
3.5 |
40 |
1.4 |
|
백혈구 감소l |
66 |
2.8 |
24 |
0 |
|
혈소판 수 감소l |
51 |
1.4 |
8 |
1.4 |
|
중성구 감소l |
42 |
2.1 |
15 |
1.4 |
|
혈액 크레아티닌 증가l,m |
19 |
0 |
12 |
0 |
a 무작위 배정된 치료로서 1회 용량 이상 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란, 각막내피결손, 각막염, 점상각막염 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질성 폐렴 포함.
e 1건의 CTCAE 5등급(치명적)이 보고됨.
f 입 궤양, 구내염 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성 발진(rash vesicular), 피부미란(skin erosion) 포함.
h 손발톱바닥장애(nail bed disorder), 손발톱바닥감염(nail bed infection), 손발톱바닥염증(nail bed inflammation), 손발톱변색(nail discolouration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
i 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부 건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k QTcF 연장이 >500 msec를 보인 환자의 발생률을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
m LAURA 시험의 베이스라인 혈액 크레아티닌 청소율(<30 mL/min)은 이 약의 다른 단독요법 시험들에서의 베이스라인 혈액 크레아틴 청소율(<50 mL/min)보다 낮으므로, 등급 변화가 일어날 가능성이 더 높았다.
표 5. FLAURA<SUP>a</SUP> 시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=279) |
EGFR TKI 대조약 (게피티닙 또는 엘로티닙) (N=277) | ||||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) | ||
|
MedDRA 선호 용어 | ||||||
|
눈 이상 | ||||||
|
각막염c |
0.4 |
0 |
1.4 |
0 | ||
|
호흡계, 흉부 및 종격 이상 | ||||||
|
비출혈 |
6.1 |
0 |
5.1 |
0 | ||
|
간질성 폐질환d |
3.9 |
1.1 |
2.2 |
1.4 | ||
|
위장관계 이상 | ||||||
|
설사e |
58 |
2.2 |
57 |
2.5 | ||
|
구내염f |
32 |
0.7 |
22 |
1.1 | ||
|
피부 및 피하조직 이상 | ||||||
|
발진g |
58 |
1.1 |
78 |
6.9 | ||
|
건성 피부h |
36 |
0.4 |
36 |
1.1 | ||
|
손발톱주위염i |
35 |
0.4 |
33 |
0.7 | ||
|
소양증j |
17 |
0.4 |
17 |
0 | ||
|
탈모증 |
7.2 |
0 |
13 |
0 | ||
|
두드러기 |
2.2 |
0.7 |
0.4 |
0 | ||
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
1.4 |
0 |
2.5 |
0 | ||
|
피부 과다 색소 침착 |
0.4 |
0 |
1.1 |
0 | ||
|
실험실적 수치 | ||||||
|
QTc 간격 연장k |
1.1 |
0.7 | ||||
|
혈액 크레아틴 인산 활성 효소 증가 |
0.4 |
0.4 | ||||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | ||||||
|
백혈구 감소l |
72 |
0.4 |
31 |
0.4 | ||
|
림프구 감소l |
63 |
5.6 |
36 |
4.2 | ||
|
혈소판수 감소l |
51 |
0.7 |
12 |
0.4 | ||
|
호중구 감소l |
41 |
3.0 |
10 |
0 | ||
|
혈액 크레아티닌 증가l |
8.8 |
0 |
6.7 |
0.4 | ||
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0.
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
e EGFR TKI 대조약 군에서 1건의 CTCAE 5등급(치명적) 사건이 보고되었다.
f 입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
i손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis), 전신 소양증(pruritis generalised) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
l실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 6. FLAURA2<SUP>a</SUP> 시험에서 보고된 이상반응
|
MedDRA SOC |
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=276) |
이 약 (N=275) | |||||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) | |||
|
MedDRA 선호 용어 | |||||||
|
눈 이상 | |||||||
|
각막염c |
0.7 |
0 |
0 |
0 | |||
|
호흡계, 흉부 및 종격 이상 | |||||||
|
비출혈 |
7 |
0.4 |
7 |
0 | |||
|
간질성 폐질환d |
3.3 |
0.7e |
3.6 |
1.8e | |||
|
위장관계 이상 | |||||||
|
설사 |
43 |
2.9 |
41 |
0.4 | |||
|
구내염f |
31 |
0.4 |
21 |
0.4 | |||
|
피부 및 피하조직 이상 | |||||||
|
발진g |
49 |
2.5 |
44 |
1.5 | |||
|
손발톱주위염h |
27 |
0.7 |
32 |
0.4 | |||
|
건성 피부i |
24 |
0 |
31 |
0 | |||
|
탈모증 |
9 |
0 |
5 |
0 | |||
|
소양증j |
8 |
0 |
11 |
0 | |||
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
5 |
0 |
3.3 |
0 | |||
|
피부 과다 색소 침착 |
2.5 |
0 |
1.1 |
0 | |||
|
두드러기 |
1.4 |
0.4 |
1.5 |
0 | |||
|
다형성 홍반 |
1.4 |
0.7 |
0.4 |
0 | |||
|
실험실적 수치 | |||||||
|
혈액 크레아틴 인산 활성 효소 증가 |
3.3 |
1.1 |
3.3 |
0 | |||
|
QTc 간격 연장k |
1.8 |
1.8 | |||||
|
(CTCAE 등급 변동으로 나타난 시험 결과에 근거한 결과) | |||||||
|
백혈구 감소l |
88 |
20 |
53 |
3.3 | |||
|
혈소판수 감소l |
85 |
16 |
44 |
1.8 | |||
|
호중구 감소l |
85 |
36 |
40 |
4.7 | |||
|
림프구 감소l |
78 |
16 |
55 |
7 | |||
|
혈액 크레아티닌 증가l |
22 |
0.4 |
8 |
0 | |||
a 무작위 배정된 치료로서 1회 용량 이상 투여 받은 환자에서 발생한 사건만 요약하였다.
b 미국 국립 암 연구소 (National Cancer Institute, NCI) 이상사례 표준 용어 기준 (Common Terminology Criteria for Adverse Events, CTCAE) 버전 5.0.
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis), 기질화 폐렴(organising pnuemonia) 포함.
e 1건의 CTCAE 5등급(치명적) 사건이 보고되었다.
f 입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 약물 발진(drug eruption), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 모낭성 발진(rash follicular) 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 소양성 발진(rash pruritic), 농포성 발진(rash pustular), 소수포성발진(rash vesicular), 피부 미란(skin erosion) 포함.
h 손발톱바닥 장애(nail bed disorder), 손발톱바닥 감염(nail bed infection), 손발톱바닥 염증(nail bed inflammation), 손발톱 변색(nail discolouration), 손발톱 장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱 감염(nail infection), 손발톱색소침착(nail pigmentation), 손발톱능성형성(nail ridging), 손발톱 독성(nail toxicity), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱 탈락(onychomadesis), 손발톱무름(onychomalacia), 손발톱주위염(paronychia) 포함.
I 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 피부건조(xeroderma), 건조증(xerosis) 포함.
j 눈꺼풀 소양증(eyelid pruritis), 소양증(pruritis) 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생률을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
표 7. AURA3<SUP>a</SUP>시험에서 보고된 이상반응
|
MedDRA SOC |
이 약 (N=279) |
항암화학요법 (페메트렉시드/시스플라틴 또는 페메트렉시드/카보플라틴) (N=136) | ||
|
NCI 등급b |
모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
MedDRA 선호용어 | ||||
|
눈 이상 | ||||
|
결막염c |
1.1 |
0 |
0.7 |
0 |
|
호흡계, 흉부 및 종격 이상 | ||||
|
비출혈 |
5.4 |
0 |
1.5 |
0 |
|
간질성 폐질환d,e |
3.6 |
0.4 |
0.7 |
0.7 |
|
위장관계 질환 | ||||
|
설사 |
41 |
1.1 |
11 |
1.5 |
|
구내염f |
19 |
0 |
15 |
1.5 |
|
피부 및 피하조직 질환 | ||||
|
발진g |
34 |
0.7 |
5.9 |
0 |
|
건성 피부h |
23 |
0 |
4.4 |
0 |
|
손발톱주위염i |
22 |
0 |
1.5 |
0 |
|
소양증j |
13 |
0 |
5.1 |
0 |
|
탈모증 |
3.6 |
0 |
2.9 |
0 |
|
두드러기 |
2.5 |
0 |
1.5 |
0 |
|
손바닥-발바닥 홍반성 감각 이상 증후군 |
1.8 |
0 |
0.7 |
0 |
|
피부 과다 색소 침착 |
0.4 |
0 |
3.7 |
0 |
|
실험실적 수치 | ||||
|
QTc 간격연장k |
1.4 |
0 | ||
|
혈액 크레아틴 인산 활성 효소 증가 |
0.7 |
0.7 | ||
|
(CTCAE grade 이동으로 나타난 시험 결과를 근거로 한 결과) | ||||
|
백혈구 감소l |
61 |
1.1 |
75 |
5.3 |
|
혈소판수 감소l |
46 |
0.7 |
48 |
7.4 |
|
호중구 감소l |
27 |
2.2 |
49 |
12 |
|
혈액 크레아티닌 증가l |
6.5 |
0 |
9.2 |
0 |
a 최소 1회 투여를 한 환자의 사례만 요약되어 있다.
b 미국 국립암연구소(National Cancer Institute, NCI) 이상사례 표준 용어 기준(Common Terminology Criteria for Adverse Events, CTCAE) 버전 4.0
c 각막미란(corneal erosion), 각막내피결손(corneal epithelium defect), 각막염(keratitis), 점상각막염(punctate keratitis) 포함.
d 간질성 폐질환(interstitial lung disease), 폐염증(pneumonitis) 포함.
e CTCAE 5등급(치명적) 1건이 보고되었다.
f입 궤양 형성(mouth ulceration), 구내염(stomatitis) 포함.
g 여드름(acne), 피부염(dermatitis), 여드름양 피부염(dermatitis acneiform), 홍반(erythema), 모낭염(folliculitis), 고름 물집(pustule), 발진(rash), 홍반성 발진(rash erythematous), 전신 발진(rash generalised), 반상 발진(rash macular), 반상‑구진 발진(rash maculo‑papular), 구진 발진(rash papular), 농포성 발진(rash pustular) 포함.
h 건성 피부(dry skin), 습진(eczema), 피부 열창(skin fissures), 건조증(xerosis) 포함.
i손발톱바닥장애(nail bed disorders), 손발톱바닥염증(nail bed inflammation), 손발톱바닥압통(nail bed tenderness), 손발톱변색(nail discoloration), 손발톱장애(nail disorder), 손발톱이영양증(nail dystrophy), 손발톱감염(nail infection), 손발톱능성형성(nail ridging), 손발톱 통증(onychalgia), 손발톱파손(onychoclasis), 손발톱박리증(onycholysis), 손발톱탈락(onychomadesis), 손발톱주위염(paronychia) 포함.
j 눈꺼풀 소양증, 소양증, 전신 소양증 포함.
k QTcF 연장 >500 msec를 보인 환자의 발생을 나타낸다.
l 실험실적 결과의 발생률을 나타낸 것으로, 보고된 이상반응이 아님.
단일군 임상2상 AURAex 및 AURA2 임상시험에서의 안전성 결과는 일반적으로 AURA3의 이 약 군에서 관찰된 것과 일치하였다. 추가적이거나 예상하지 못한 독성이 관찰되지 않았으며, 이상반응은 종류, 중증도 및 빈도 등이 알려진 것과 유사하였다.
선택된 이상반응에 대한 설명
혈액학적 독성
이 약으로 치료를 받은 환자에서 실험실적 백혈구, 림프구, 호중구 및 혈소판 수의 중앙값에 조기 감소가 관찰되었으며, 이는 시간이 지남에 따라 안정해진 후 정상 하한보다 높게 유지되었다. 백혈구 감소증, 림프구 감소증, 호중구 감소증, 및 혈소판 감소증 이상사례가 보고되었으며, 대부분은 경증 또는 중등증이었고 투여 중지로 이어지지 않았다.
3) 국내 시판 후 수집된 중대한 이상사례 분석▪평가 결과 확인된 이상사례는 다음과 같다. 다만, 이로써 곧 해당성분과 다음의 이상사례 간에 인과관계가 입증된 것을 의미하는 것은 아니다.
◦ 호흡기계 : 폐색전증
※ 국내 시판 후 조사 결과
국내에서 1178명을 대상으로 실시한 시판 후 조사 결과, 이상사례의 발현율은 인과관계와 상관없이 69.10%(814/1178명, 2287건)로 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응 및 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 발현 빈도에 따라 아래 표에 나열하였다.
|
발현빈도 |
기관계 |
중대한 약물이상반응 6.79% (80/1178명, 99건) |
예상하지 못한 약물이상반응 17.74% (209/1178명, 296건) |
|
흔하게 (1~10% 미만) |
임상 검사 |
- |
알라닌 아미노 전이 효소 증가, 아스파르트산 아미노 전이 효소 증가 |
|
각종 위장관 장애 |
- |
변비 | |
|
대사 및 영양 장애 |
- |
식욕 감소 | |
|
전신 장애 및 투여 부위 병태 |
- |
점막 염증 | |
|
흔하지 않게 (0.1~1% 미만) |
호흡기, 흉곽 및 종격 장애 |
폐염증, 간질성 폐 질환, 폐 색전증 |
호흡 곤란, 기침, 흉막 삼출 |
|
혈액 및 림프계 장애 |
중성구 감소증, 혈소판 감소증 |
빈혈 | |
|
각종 위장관 장애 |
구역, 설사, 복통 |
구토, 소화 불량, 복통, 상복부 통증, 입 건조, 식도염, 위염 | |
|
감염 및 기생충 감염 |
폐렴 |
폐렴 | |
|
신장 및 요로 장애 |
급성 신 손상 |
급성 신 손상 | |
|
피부 및 피하 조직 장애 |
약물 발진 |
피부 병변, 피부 탈락, 피부 반응 | |
|
전신 장애 및 투여 부위 병태 |
통증 |
무력증, 피로, 말초 부종, 통증, 전신 부종 | |
|
간담도 장애 |
간염, 약물-유발 간 손상 |
간염, 약물-유발 간 손상 | |
|
대사 및 영양 장애 |
- |
섭식 저하 | |
|
각종 신경계 장애 |
- |
말초 신경 병증, 감각 저하, 어지러움, 두통, 지각 이상 | |
|
각종 심장 장애 |
- |
심장막 삼출 | |
|
근골격 및 결합 조직 장애 |
- |
관절통 | |
|
각종 눈 장애 |
- |
눈 건조 | |
|
드물게 (0.01~0.1% 미만) |
호흡기, 흉곽 및 종격 장애 |
호흡 곤란, 흉막 삼출 |
구인두 통증, 폐 침윤 |
|
혈액 및 림프계 장애 |
발열성 중성구 감소증, 백혈구 감소증 |
발열성 중성구 감소증, 백혈구증 | |
|
각종 위장관 장애 |
구토, 흑색변 |
위 식도 역류 질환, 위 궤양, 흑색변, 혀 발진, 혀 궤양 형성 | |
|
감염 및 기생충 감염 |
요로 감염, 충수염, 클로스트리듐 디피실레 결장염, 위장염 |
요로 감염, 위장염, 구강 칸디다증, 부비동염, 충수염, 클로스트리듐 디피실레 결장염, 림프선 감염 | |
|
각종 심장 장애 |
급성 심근 경색, 심부전, 심근 병증, 심독성, 확장 심근 병증, 두근거림, 심장막 삼출 |
심부전, 두근거림, 급성 심근 경색, 심근 병증, 심독성, 확장 심근 병증, 심실 위 빈맥 | |
|
신장 및 요로 장애 |
만성 신장병, 신부전, 신 기능 장애 |
야간뇨, 만성 신장병, 신부전, 신 기능 장애 | |
|
피부 및 피하 조직 장애 |
피부 혈관염, 피부염, 발진 |
수포, 모낭 장애, 손발톱 소피 균열, 건선, 딱지, 피부 삼출 | |
|
각종 신경계 장애 |
어지러움, 뇌 병증, 말초 신경 병증, 신경 독성 |
진전, 당뇨 신경 병증, 신경통, 말초 감각 신경 병증, 뇌 병증, 신경 독성 | |
|
전신 장애 및 투여 부위 병태 |
무력증, 비-심장성 흉통 |
발열, 안면 부종, 부종, 국소 부종, 비-심장성 흉통 | |
|
대사 및 영양 장애 |
식욕 감소, 당뇨병, 고칼슘 혈증 |
당뇨병, 고칼슘 혈증 | |
|
양성, 악성 및 상세 불명의 신생물(낭종 및 용종 포함) |
위장관 선종, 악성 흉막삼출액 |
위장관 선종, 악성 흉막삼출액 | |
|
임상 검사 |
심전도 QT 연장, 중성구 수 감소 |
혈액 알칼리 인산 분해 효소 증가, 간 효소 증가 | |
|
각종 혈관 장애 |
말초 동맥 협착 |
열감 홍종, 말초 동맥 협착 | |
|
각종 내분비 장애 |
부신 부전 |
부신 부전 | |
|
근골격 및 결합 조직 장애 |
- |
근육통, 근육 쇠약, 옆구리 통증, 관절 주위염, 근육 연축, 관절염 | |
|
각종 눈 장애 |
- |
백내장, 시각 장애, 시야 흐림, 눈 분비물 | |
|
생식계 및 유방 장애 |
- |
질 출혈, 자궁 경부 용종, 월경 사이 출혈 |
각종 신경계 장애 ‑ 미각 이상
근골격 및 결합 조직 장애 ‑ 등허리 통증, 사지 통증, 근육 연축
대사 및 영양 장애 ‑ 식욕 감소
전신 장애 및 투여 부위 병태 ‑ 무력증, 피로
4. 일반적 주의
1) EGFR 변이 상태 및 검사
비소세포폐암 환자에서 완전 종양 절제술 후 보조 치료로서 이 약의 사용을 고려할 때, EGFR 변이 양성 상태 (엑손 19 결손 또는 엑손 21[L858R] 치환 변이)는 치료 적격성을 나타낸다.
이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
절제 불가능한 국소 진행성(III기) 비소세포폐암 환자에서 이 약의 사용을 고려할 때, EGFR 돌연변이 양성 상태(엑손 19 결손 또는 엑손 21 [L858R] 치환 변이)는 치료 적격성을 나타낸다. 이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
국소 진행성 또는 전이성 비소세포폐암의 치료제로서 이 약의 사용을 고려할 때, 이러한 기술의 사용이 능숙한 실험실에서 EGFR 변이 양성 상태 (1차 치료: 엑손 19 결손 또는 엑손 21[L858R] 치환 변이, 이전에 EGFR‑TKI로 치료 받은 적이 있는 환자: T790M 변이)를 확인하는 것이 중요하다.
단독요법에서 보조 치료 임상시험에서는 EGFR 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였다.
단독요법 및 페메트렉시드와 백금 기반 항암화학요법과 병용 요법의 1차 치료 임상시험에서는 EGFR 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였고, 인증받은 지역 실험실 검사도 사용되었다.
임상시험에서는 T790M 변이 양성 비소세포폐암을 진단하기 위해, 로슈진단(주)의 Cobas® EGFR Mutation Test를 이용하였다.
2) 이 약은 운전 및 기계 조작하는 능력에 영향을 미치지 않거나 또는 현저한 영향을 미치지 않는다.
5. 상호작용
1) 이 약의 혈장 농도를 증가시킬 수 있는 활성 물질
생체 외(in vitro) 시험에서 이 약의 1단계 대사는 주로 CYP3A4 및 CYP3A5를 통해 일어나는 것으로 확인되었다. 약동학적 임상시험에서, 이 약과 이트라코나졸(강력한 CYP3A4 저해제) 1일 2회 200mg를 병용투여 하였을 때 이 약의 노출(커브아래면적(AUC;Area Under the Curve)은 24%(90% CI 15, 35) 증가하였고 C<SUB>max</SUB>는 ‑20%(90% CI ‑27, ‑13) 감소)에는 임상적으로 유의한 영향을 미치지 않았다. 따라서 CYP3A4 저해제는 이 약의 노출량에 영향을 미치지 않는 것으로 보인다.
2) 이 약의 혈장 농도를 감소시킬 수 있는 활성 물질
강력한 CYP3A4 유도제는 오시머티닙의 노출을 감소시킬 수 있다. 환자를 대상으로 한 약동학적 임상시험에서, 리팜피신(21일간 600mg매일 투여)과 병용투여 시 이 약의 정상상태의 AUC는 ‑78%(90% CI ‑81, ‑76) 감소하였다. 이 약과 강력한 CYP3A 유도제(예, 페니토인, 리팜피신, 카바마제핀, 세인트존스워트(St. John’s Wort))와의 병용투여는 피하는 것이 권장된다.
3) 위산을 감소시키는 활성 성분이 이 약의 대사 미치는 영향
환자를 대상으로 한 약동학적 임상시험에서, 오메프라졸과의 병용투여는 이 약의 노출량에 대하여 임상적으로 관련 있는 변화를 야기하지 않았다. 위내 pH를 변화시키는 약물들은 제한 없이 이 약과 병용투여 할 수 있다.
4) 이 약에 의해 혈장 농도가 변할 수 있는 활성 성분
생체 외 시험에 근거하였을 때, 이 약은 BCRP 수송체의 경쟁적 저해제이다. 이 약은 breast cancer resistant protein(BCRP) 및 P‑gp 기질의 노출을 증가시킬 수 있다.
환자를 대상으로 한 약동학적 임상시험에서, 이 약과 로수바스타틴(민감한 BCRP 기질)의 병용투여는 로수바스타틴의 AUC와 C<SUB>max</SUB>를 각각 35%(90% CI 15, 57), 72%(90% CI 46, 103) 증가시켰다.
BCRP에 약물 분포가 의존적이며 치료영역이 좁은 약물을 병용 투여하는 환자는 초기용량 설정 시 약동학적 상호작용 가능성을 면밀히 고려하고 이 약 투여 중 병용약물의 노출량 증가에 기인하여 발생할 수 있는 내약성 변화의 징후에 대해 주의 깊게 모니터링 해야 한다.
환자를 대상으로 한 약동학적 임상시험에서, 이 약과 심바스타틴(민감한 CYP3A4 기질)의 병용투여는 심바스타틴의 AUC와 C<SUB>max</SUB>를 각각 ‑9% (90% CI ‑23, 8), ‑23 % (90% CI ‑37, ‑6) 감소시켰다. 이 변화는 작고, 임상적으로 유의하지 않은 것으로 보인다. CYP3A4 기질과의 임상적 약동학적 상호작용은 없는 것으로 보인다.
임상 약동학 시험에서, 이 약과와 펙소페나딘(PXR/P‑gp 기질)의 병용 투여는 펙소페나딘의 AUC 및 C<SUB>max</SUB>를 단회 투여 후 각각 56% (90% CI 35, 79) 및 76% (90% CI 49, 108) 증가시켰고 항정상태에서 각각 27% (90% CI 11, 46) 및 25% (90% CI 6, 48) 증가시켰다. P‑gp에 약물 분포가 의존적이며 치료 영역이 좁은 약물(예, 디곡신, 다비가트란, 알리스키렌)을 병용 투여하는 환자는 이 약 투여 중 병용 약물의 노출 증가에 기인한 내약성 변화의 징후에 대해 주의 깊게 모니터링 되어야 한다.
6. 임부 및 수유부에 대한 투여
1) 남성 및 여성에서의 피임
가임 여성은 이 약 투여 중 임신을 피하여야 한다. 환자는 이 약 투여 완료 후 여성은 최소 2개월, 남성은 최소 4개월 동안 효과적인 피임법을 계속 사용하도록 해야 한다.
2) 임부
이 약을 임부에게 투여한 자료는 없거나 제한적이다. 동물시험에서는 생식독성을 보였다.
작용기전 및 비임상시험 데이터에 근거할 때, 이 약은 임신한 여성에게 투여 시 태아 독성을 유발할 수 있다. 임신한 랫드에게 오시머티닙 투여 시, 사람에게 예상되는 노출량과 유사한 수준에서 배자 치사, 태자 성장 감소 및 신생자(neonate) 사망과 관련이 있는 것으로 나타났다. 이 약은 임부 및 피임을 하지 않고 임신할 가능성이 있는 여성에게 투여하지 않는다.
3) 수유부
오시머티닙 또는 그 대사체가 사람 모유로 분비되는지는 알려져 있지 않다.
임신한 랫드 및 초기 수유 중인 랫드에의 이 약의 투여는 성장율 감소와 신생자 사망(neonatal death)을 포함한 이상반응과 관련이 있었다. 이 약 또는 그 대사체가 동물의 모유로 분비되는지에 대한 정보는 충분하지 않다. 젖먹이 아이에게의 위험을 배제할 수 없다. 수유부에게 이 약 치료 중 수유를 중단하도록 해야 한다.
4) 수태능
사람의 수태능에 이 약이 미치는 영향에 대한 자료는 없다. 동물실험 결과 이 약이 남성 및 여성 생식기관에 영향을 주어 생식능력을 저하시킬 수 있는 것으로 나타났다.
7. 소아에 대한 투여
소아 환자에서 이 약의 안전성 ◦유효성은 확립되지 않았다.
8. 고령자에 대한 투여
집단 약동학 분석에서 연령은 오시머티닙의 노출량에 영향을 주지 않는 것으로 나타났다.
ADAURA, FLAURA, FLAURA2 및 AURA (이 약의 단독요법 군 (N = 1813))에서, 42 % 환자가 65세 이상이었고, 11%가 75세 이상이었다. 젊은 환자(65세 미만)와 비교하여, 65세 이상의 환자에서 시험약 용량 변경이 필요한 이상반응이 더 많이 보고되었다(일시중단 또는 용량감소)(14% vs 10%). 보고된 이상반응의 종류는 연령에 관계없이 유사하였다. 고령의 환자가 젊은 환자에 비해 3등급 이상의 이상반응을 더 많이 보고하였다(11% vs 9%). 고령의 환자와 젊은 환자 간 유효성에 전반적인 차이는 관찰되지 않았다.
9. 간장애 환자에 대한 투여
임상시험에 근거할 때 경증(Child Pugh A) 또는 중등증 간장애(Child Pugh B) 환자에서의 용량 조절은 필요하지 않다. 이와 유사하게, 집단 약동학 분석에 근거할 때, 경증의 간장애 환자(총빌리루빈≦정상 상한치[ULN] 이고 AST >ULN 또는 총빌리루빈이 1.0배의 ULN과 1.5배의 ULN 및 모든 수치의 AST 사이) 또는 중등증의 간장애 환자(총빌리루빈이 1.5배의 ULN과 3배의 ULN 및 모든 수치의 AST 사이)에서 용량 조절은 필요하지 않다.
중증의 간장애 환자에서 이 약의 안전성 ◦유효성은 확립되지 않았다.
10. 신장애 환자에 대한 투여
임상시험 및 집단 약동학 분석에 근거할 때, 경증, 중등증 또는 중증의 신장애 환자에게 용량조절은 필요하지 않다.
말기 신질환 환자 [Cockcroft and Gault 방정식에 의해 계산된 크레아티닌 청소율 (CLcr)이 15 mL/min 미만] 또는 투석 중인 환자에 대한 이 약의 안전성 ◦유효성은 확립되지 않았다. 중증 및 말기 신장애 환자 치료 시 주의해야 한다.
11. 과량투여시의 처치
이 약을 사용한 임상시험에서 제한된 수의 환자들이 이 약 1일 최대 240mg까지 용량 제한적 독성(dose limiting toxicities)없이 투여되었다. 이 연구들에서, 이 약을 1일 160mg 및 240mg 투여받은 환자들은 80mg을 투여받은 환자들보다 전형적인 EGFR‑유도 이상반응(주로 설사와 피부발진)의 빈도 및 중증도의 증가를 경험하였다.
이 약의 과량투여 시 특별한 치료방법이 없다. 의사는 일반적인 보조 조치를 실시하고 대증적으로 치료해야 한다.
12. 보관 및 취급상의 주의사항
1) 어린이의 손이 닿지 않는 곳에 보관한다.
2) 다른 용기에 바꾸어 넣는 것은 사고원인이 되거나 품질 유지 면에서 바람직하지 않으므로 이를 주의한다.
13. 전문가를 위한 정보
1) 약리작용
이 약은 티로신 키나제 억제제 (Tyrosine Kinase Inhibitor, TKI)이다. 이 약은 감작 돌연변이 (EGFRm) 및 TKI‑내성 돌연변이 T790M을 가진 표피 성장인자 수용체 (EGFRs)의 강력한 선택적 비가역적 경구 억제제이다.
In vitro 시험에서 이 약은 임상적으로 관련된 모든 EGFR 감작 돌연변이체 및 T790M 돌연변이체 비소세포폐암 (NSCLC) 다양한 세포주에 대하여 높은 효력 및 억제 활성을 나타내는 것을 입증하였다. (phospho‑EGFR에 대한 겉보기 IC<SUB>50</SUB>6 nM에서 54 nM) 이로 인해 세포 성장이 억제되는 반면, 야생형 세포주에서는 EGFR에 대한 활성이 유의하게 낮게 나타난다. (phospho‑EGFR에 대한 겉보기 IC<SUB>50</SUB>480 nM에서 1.9 μM) In vivo에서 이 약의 경구 투여는 EGFRm 및 T790M 모두의 NSCLC 이종이식 및 유전자 이식 마우스 폐 종양 모델에서 종양 축소를 나타난다.
심장 전기생리학
이 약의 QTc 간격 연장 가능성은 AURA2에서 오시머티닙 1일 80 mg을 투여 받은 210 명의 환자에서 평가되었다. QTc 간격에 대한 오시머티닙의 영향을 평가하기 위해 연속적 ECG를 단회 투여 및 정상상태에서 수집했다. 이 약의 약동학/약력학 분석은 80 mg에서 14 msec의 약물 관련 QTc 간격 연장을 상한 16 msec (90% CI)으로 예측했다.
2) 약동학적 정보
오시머티닙 약동학 파라미터는 건강한 시험대상자 및 NSCLC 환자들에서 확인되었다. 집단 약동학 분석에 근거하여, 오시머티닙 겉보기 혈장 청소율은 14.3 L/h이며, 겉보기 분포 용적은 918 L이고, 말단 반감기는 약 44시간이다. 이 약과 페메트렉시드 및 백금 기반 항암화학요법을 병용하여 치료한 환자의 약동학은 이 약 단독요법으로 치료받은 환자의 약물동태와 유사하다. AUC 및 Cmax 는 20에서 240mg 용량 범위에서 용량에 비례하여 증가하였다. 이 약을 1일 1회 투여한 결과, 약 3배의 축적을 나타내며, 정상 상태 노출은 투여 15일에 도달되었다. 정상 상태에서, 순환혈장농도는 일반적으로 24시간 투여 간격 동안 1.6배 범위 내에서 유지된다.
흡수
이 약을 경구 투여한 후, 오시머티닙의 최고 혈장 농도는 t<SUB>max</SUB>중앙값(최소‑최대)은 6시간 (3‑24)에 도달되었으며, 일부 환자들에서 처음 24시간 동안 여러 개의 피크가 관찰되었다. 이 약의 절대 생체 이용률은 70%이다. (90% CI 67, 73) 80 mg을 투여 받은 환자에 대한 임상 약동학 시험에 근거하여, 음식은 오시머티닙의 생체 이용률을 임상적으로 의미 있는 정도로 변경하지 않는다. (AUC 6% 증가 (90% CI ‑5, 19) 및 C<SUB>max</SUB>‑7%감소 (90% CI ‑19, 6)). 5일간 오메프라졸 투여에 의해 위장 pH가 상승한 건강한 시험대상자에서 80 mg 정제를 투여했을 때, 오시머티닙 노출은 영향을 받지 않았으며 (AUC 및 C<SUB>max</SUB>증가 각각 7% 및 2%), 노출비에 대한 90% CI는 80‑125% 한도 내에 포함되었다.
분포
모집단에서 평가된 오시머티닙의 정상 상태에서의 평균 분포 용적은 (V<SUB>ss</SUB>/F)918L이며, 이는 조직으로의 광범위한 분포를 시사한다. In vitro에서, 오시머티닙의 혈장 단백질 결합은 94.7%이다. (비결합 5.3%) 오시머티닙은 또한 랫드와 사람의 혈장 단백질, 사람 혈청 알부민 및 랫드와 사람 간세포에 공유 결합하는 것으로 확인되었다.
생체 내 변환
In vitro 시험에서 오시머티닙은 주로 CYP3A4 및 CYP3A5에 의해 대사되는 것을 보여준다. In vitro 시험에 근거하여, 두가지 약리학적 활성 대사체 (AZ7550 및 AZ5104)가 이 약을 경구 투여한 후 전임상 종의 혈장 및 사람에서 이후 확인되었다; AZ7550은 이 약과 유사한 약리학적 프로파일을 나타낸 반면, AZ5104는 돌연변이 및 야생형 EGFR 모두에서 더 큰 효력을 보였다. 환자들에게 이 약을 투여한 후 두 대사체 모두 혈장에서 서서히 나타났으며, t<SUB>max</SUB>중앙값 (최소‑최대)은 각각 24 (4‑72) 및 24 (6‑72) 시간이었다. 사람 혈장에서, 모성분 오시머티닙은 0.8%를 차지하였고, 두 대사체는 총 방사능의 0.08% 및 0.07%에 기여하였으며, 방사능의 대부분은 혈장 단백질에 공유 결합하였다. AUC에 근거한 AZ5104 및 AZ7550의 기하 평균 노출은 정상 상태에서 오시머티닙 노출의 각각 약 10%였다.
오시머티닙의 주요 대사 경로는 산화 및 탈알킬화였다. 사람의 뇨 및 분변 통합 검체에서 최소 12개의 성분들이 관찰되었으며, 5개 성분은 투여량의 >1%를 차지하였고, 이 중 미변화 오시머티닙, AZ5104, 및 AZ7550은 투여량의 약 1.9, 6.6, 및 2.7%를 차지한 반면, cysteinyl 부가생성물 (M21), 및 알려지지 않은 대사체 (M25)는 투여량의 각각 1.5% 및 1.9%를 차지하였다.
In vitro 시험에 근거하여 오시머티닙은 임상적으로 관련된 농도에서 CYP 3A4/5의 경쟁적 억제제이지만, CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 및 2E1 에 대해서는 아니다. In vitro 시험에 근거하여 오시머티닙은 간에서 임상적으로 관련된 농도에서 UGT1A1 및 UGT2B7의 억제제가 아니다. UGT1A1의 장에서의 억제는 가능하지만, 임상적 영향은 알려지지 않았다.
배설
20 mg 단회 경구 투여 후, 투여량의 67.8%가 분변으로 회수된 반면 (모성분으로 1.2%), 투여량의 14.2% (모성분으로 0.8%)는 84일의 표본 수집 시까지 소변에서 발견되었다. 미변화된 오시머티닙은 배설의 약 2%를 차지하였으며, 0.8%는 소변으로, 1.2%는 분변으로 배설되었다.
특수 집단
집단 기반 PK 분석에서 (n=1367), 예측된 정상 상태 노출 (AUC<SUB>ss</SUB>)과 환자의 연령 (범위: 25‑91세), 성별 (65% 여성), 인종 (백인, 아시아인, 일본인, 중국인 및 비아시아‑비백인 환자), 치료 차수 및 흡연 상태 (n=34 현재 흡연자, n=419 과거 흡연자) 간에 임상적으로 유의한 관련성은 확인되지 않았다. 집단 PK 분석에서 체중이 유의한 공변량으로 나타났으며, 체중 중앙값 61 kg의 AUC<SUB>ss</SUB>와 비교했을 때, 88 kg에서 43 kg 체중 범위에서 오시머티닙 AUC<SUB>ss</SUB>변화가 20% 미만으로 예상된다. (95%‑5% 사분위) 체중의 극단을 고려하여 43 kg 미만에서 88 kg 초과까지 AZ5104 대사체 비는 11.8% 에서 9.6% 범위이며, AZ7550의 경우 그 범위는 12.8%에서 8.1%이다. 집단 PK 분석에 근거하여 혈청 알부민은 유의한 공변량으로 확인되었으며, 베이스라인 알부민 중앙값 39 g/L에 대한 AUC<SUB>ss</SUB>와 비교했을 때 알부민 범위 29에서 46 g/L의 알부민 범위에서 오시머티닙 AUCss 변화는 30% 미만으로 예상된다. (95% ‑ 5% 사분위수) 체중 또는 베이스라인 알부민 차이에 의한 이러한 노출 변화는 임상적으로 관련이 없는 것으로 고려된다.
뇌 전이 환자
뇌 전이가 있는 EGFR 돌연변이 양성 NSCLC 환자 (n=4)를 대상으로 한 마이크로도즈 PET 연구에서 오시머티닙의 뇌 침투 및 분포(중앙값 T<SUB>max</SUB>: 22분;평균C<SUB>max</SUB>: 투여된 용량의 1.5%가 뇌에 도달)는 건강한 지원자 연구에서 관찰된 것과 유사했다 (n=7; 중앙값 T<SUB>max</SUB>: 11분; 평균 C<SUB>max</SUB>: 투여된 용량의 2.2%가 뇌에 도달).
3) 임상적 유효성 및 안전성
이전 보조 화학 요법을 받았거나 받지 않은 EGFR 변이 양성 비소세포폐암의 보조 치료 ‑ ADAURA
이전 보조 화학 요법을 받았거나 받지 않은 완전 종양 절제술을 받은 EGFR 변이 양성 (엑손 19 결손 또는 L858R 치환 변이) 비소세포폐암 환자의 보조 치료에 대한 이 약의 유효성 및 안전성은 무작위 이중 맹검 위약 대조 시험 (ADAURA)에서 입증되었다.
절제 가능한 종양 (IA기 제외)이 있는 환자들은 중앙 실험실에서 생검 또는 수술 검체를 사용하여 전향적으로 실시된 cobas EGFR Mutation Test에 의해 확인된 EGFR 엑손 19 결손 또는 엑손 21 L858R 치환 변이가 있어야 했다.
환자들은 무작위 배정되어 (1:1) 이 약 80 mg 경구 1일 1 회 또는 위약을 수술에서 회복된 후 투여 받고 제공된 경우 표준 보조 화학 요법을 받았다. 보조 화학 요법을 받지 않은 환자들은 수술 후 10 주 이내에, 보조 화학 요법을 받은 환자들은 수술 후 26 주 이내에 무작위 배정되었다. 무작위 배정은 돌연변이 유형 (엑손 19 결손 또는 엑손 21 L858R 치환 변이), 민족성 (아시아인 또는 비아시아인) 및 AJCC 7판에 따른 pTNM (IB 또는 II 또는 IIIA) 기반 병기 별로 계층화되었다. 질병이 재발하거나 수용할 수 없는 독성이 나타날 때까지 또는 3년 동안 투여를 받았다.
주요 유효성 결과 측정은 연구자 평가에 따른 무병 생존 기간 (disease‑free survival, DFS)이었다. 추가적 유효성 결과 측정은 DFS 비율, 전체 생존기간 (overall survival, OS), OS 비율 및 건강 관련 삶의 질 (HRQL) SF‑36이 악화되는 시간을 포함한다.
총 682 명의 환자가 이 약 (n = 339) 또는 위약 (n = 343)에 무작위 배정되었다. 중앙 연령은 63세 (30‑86세 범위)였고 11%는 75세 이상; 70%는 여성, 64%는 아시아인, 72%는 흡연 미경험자였다. 베이스라인 WHO 수행 능력은 0 (64%) 또는 1 (36%)이었다; 31%는 IB기, 34% II기, 그리고 35%는 IIIA기였다. EGFR 돌연변이 상태와 관련하여, 55%는 엑손 19 결손이었고 45%는 엑손 21 L858R 치환 변이였다; 9명의 환자 (1%)는 de novo T790M 변이도 있었다. 환자의 대다수 (60%)는 무작위 배정 전 보조 화학 요법을 받았다 (26% IB; 71% IIA; 73% IIB; 80% IIIA).
II‑IIIA기 집단과 전체 집단 (IB‑IIIA)에 대해 DFS 분석을 했다. ADAURA는 위약으로 치료받은 환자 대비 이 약으로 치료받은 환자에 대해 질병 재발 또는 사망의 위험이 통계적으로 유의하고 임상적으로 의미있는 감소함을 입증하였다. 이 약으로 치료받은 II‑IIIA기 질환 환자들은 위약 대비 질병 재발 또는 사망의 위험이 83% 감소했다 (중앙값은 각각 계산되지 않음 (not calculated, NC) 및 19.6개월, HR=0.17, 99.06% CI:0.11, 0.26; P<0.0001). 위약 대비 이 약으로 치료 받은 전체 집단 (IB‑IIIA)은 질병 재발 또는 사망의 위험이 80% 감소한 것으로 나타났다 (중앙값은 각각 NC 및 27.5개월, HR=0.20, 99.12% CI:0.14, 0.30; P<0.0001).
이 약에서 질병이 재발한 환자는 37명이었다. 가장 흔하게 보고된 재발 부위는 다음과 같다; 폐 (19 명); 림프절 (10명) 및 CNS (5명). 위약으로 질병이 재발한 환자는 157명이었다. 가장 흔하게 보고된 부위는 다음과 같다; 폐 (61명); 림프절 (48명) 및 CNS (34명).
OS의 최종 분석은 II‑IIIA기 집단 (21.3% 성숙; HR=0.49; 95.03% CI: 0.33, 0.73; p‑값=0.0004) 및 전체 집단 (IB‑IIIA; 18.2% 성숙; HR=0.49; 95.03% CI: 0.34, 0.70; p 값 < 0.0001) 둘 모두에 대해 위약과 비교하여 이 약을 투여받은 환자의 51% 사망 위험 감소를 보이며 임상적으로 의미 있고 통계적으로 크게 유의한 OS 개선을 입증하였다. 전체 집단 (IB‑IIIA)에서, 중도절단된 환자의 추적관찰 시간의 중앙값은 두 치료군 모두 61.5개월이었다.
연구자 평가에 따른 ADAURA의 유효성 결과는 표 8 및 9에 요약되어 있으며, II‑IIIA기 환자 및 전체 집단 (IB‑IIIA)의 DFS 및 OS 에 대한 Kaplan‑Meier 곡선은 그림 1부터 그림 4에 제시하였다.
표 8. 연구자 평가에 따른 II‑IIIA기 환자에서의 유효성 결과
|
유효성 지표 |
이 약 (N=233) |
위약 (N=237) |
|
무병 생존 기간 (Disease Free Survival) | ||
|
사건 수 (%) |
26 (11.2) |
130 (54.9) |
|
질병 재발 (%) |
26 (11.2) |
129 (54.4) |
|
사망 (%) |
0 |
1 (0.4) |
|
DFS 중앙값, 개월(95% CI) |
NC (38.8, NC) |
19.6 (16.6, 24.5) |
|
HR (99.06% CI); P-값a |
0.17 (0.11, 0.26); <0.0001 | |
|
12개월 시점 DFS 비율 (%) (95% CI) |
97.2 (93.9, 98.7) |
60.8 (54.1, 66.8) |
|
24개월 시점 DFS 비율 (%) (95% CI) |
89.5 (84, 93.2) |
43.6 (36.5, 50.6) |
|
36개월 시점 DFS 비율 (%) (95% CI)b |
78.3 (64.5, 87.3) |
27.9 (18.9, 37.6) |
|
전체 생존 기간 (Overall Survival) | ||
|
사망 수 (21% 성숙) |
35 (15.0) |
65 (27.4) |
|
OS 중앙값, 개월 (95% CI) |
NC (NC, NC) |
NC (NC, NC) |
|
HR (95.03% CI); P-값c |
0.49 (0.33, 0.73); 0.0004 | |
|
24개월 시점 OS 비율 (%) (95% CI)d |
99.5 (96.8, 99.9) |
92.6 (88.3, 95.3) |
|
36개월 시점 OS 비율 (%) (95% CI)d |
94.1 (90.1, 96.5) |
85.9 (80.6, 89.8) |
|
48개월 시점 OS 비율 (%) (95% CI)d |
91.0 (86.3, 94.1) |
79.9 (74.0, 84.6) |
|
60개월 시점 OS 비율 (%) (95% CI)d |
85.0 (79.3, 89.2) |
72.6 (66.0, 78.1) |
DFS 결과는 연구자 평가에 근거한 것이다.
HR <1은 이 약에 유리하다.
DFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 22.1개월이었고 위약을 투여 받은 환자에서 14.9개월이었다.
중도절단된 환자의 OS에 대한 추적관찰 시간의 중앙값은 이 약 투여 군(stage II‑IIIA 집단)에서 61.7개월, 위약 군에서 60.4개월이었다.
DFS 결과는 1차 분석(2020년 1월 17일)에서 나왔다. OS 결과는 최종 분석(2023년 1월 27일)에서 나왔다.
a 중간분석 (33% 성숙)을 위해 조정된 p‑값 <0.0094이 통계적 유의성을 달성하기 위해 필요했다.
b 36개월 시점에 추적 관찰 환자 (patient at risk) 수는 이 약 투여군은 18명이고, 위약 군은 9명이었다.
c 중간 분석을 위해 조정된 통계적 유의성을 달성하려면 p‑값 <0.0497이 필요했다.
d Kaplan‑Meier 방법으로 계산했다.
그림 1. 연구자 평가에 따른 무병 생존 기간 (II‑IIIA기 환자)의 Kaplan‑Meier 곡선
|
유효성 지표 |
이 약 (N=339) |
위약 (N=343) |
|
무병 생존 기간 (Disease Free Survival) | ||
|
사건 수 (%) |
37 (10.9) |
159 (46.4) |
|
질병 재발 (%) |
37 (10.9) |
157 (45.8) |
|
사망 (%) |
0 |
2 (0.6) |
|
DFS 중앙값, 개월 (95% CI) |
NC (NC, NC) |
27.5 (22.0, 35.0) |
|
HR (99.12% CI); P -값a |
0.20 (0.14, 0.30); <0.0001 | |
|
12개월 시점 DFS 비율 (%) (95% CI) |
97.4 (94.9, 98.7) |
68.5 (63.2, 73.2) |
|
24개월 시점 DFS 비율 (%) (95% CI) |
89.1 (84.5, 92.4) |
52.4 (46.4, 58.1) |
|
36개월 시점 DFS 비율 (%) (95% CI)b |
78.9 (68.7, 86.1) |
40.0 (32.1, 47.8) |
|
전체 생존 기간 (Overall Survival) | ||
|
사망 수 (18% 성숙) |
42 (12.4) |
82 (23.9) |
|
OS 중앙값, 개월 (95% CI) |
NC (NC, NC) |
NC (NC, NC) |
|
HR (95.03% CI); P-값c |
0.49 (0.34, 0.70); <0.0001 | |
|
24개월 시점 OS 비율 (%) (95% CI)d |
99.4 (97.5, 99.8) |
94.3 (91.2, 96.3) |
|
36개월 시점 OS 비율 (%) (95% CI)d |
95.3 (92.3, 97.1) |
88.8 (84.9, 91.8) |
|
48개월 시점 OS 비율 (%) (95% CI)d |
92.8 (89.4, 95.2) |
83.9 (79.4, 87.4) |
|
60개월 시점 OS 비율 (%) (95% CI)d |
87.6 (83.3, 90.9) |
77.7 (72.7, 81.9) |
HR (Hazard Ratio)=위험비; CI (Confidence Interval)=신뢰 구간; NC (Not Calculable)=계산 불가능
DFS 결과는 연구자 평가에 근거한 것이다.
HR <1은 이 약에 유리하다.
DFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 22.1개월이었고 위약을 투여 받은 환자에서 16.6개월이었다.
중도절단된 환자의 OS에 대한 추적관찰 시간의 중앙값은 이 약 투여 군 및 위약 군에서 모두 61.5개월이었다.
DFS 결과는 1차 분석(2020년 1월 17일)에서 나왔다. OS 결과는 최종 분석(2023년 1월 27일)에서 나왔다.
a 중간분석 (29% 성숙)을 위해 조정된 p‑값 < 0.0088이 통계적 유의성을 달성하기 위해 필요했다.
b 36개월 시점에 추적 관찰 환자 (patient at risk) 수는 이 약 투여 군은 27명이고, 위약 군은 20명이었다.
c 중간 분석을 위해 조정된 통계적 유의성을 달성하려면 p‑값 < 0.0497이 필요했다.
d Kaplan‑Meier 방법으로 계산했다.
그림 3. 연구자 평가에 따른 무병 생존 기간 (전체 집단)의 Kaplan‑Meier 곡선
위약 환자 대비 이 약 환자에 대한 CNS DFS (CNS 재발 또는 사망까지의 시간)의 탐색적 분석에서 임상적으로 의미있는 개선이 전체 집단에서 HR 0.18 (95% CI:0.10, 0.33; P <0.0001)로 관찰되었으며, 이는 위약 대비 이 약 군에서 CNS 질환 재발 또는 사망의 위험이 82% 감소함을 나타낸다.
환자 보고 결과
ADAURA에서 건강 관련 삶의 질 (HRQL)은 Short Form (36) Health Survey version 2 (SF‑36v2) 설문지를 사용하여 평가되었다. SF‑36v2는 치료가 완료되거나 중단될 시까지 무작위 배정과 관련하여 12주, 24주, 그 후 24주마다 관리되었다. 전반적으로, HRQL은 두 군 모두에서 유지되었으며, II‑IIIA기 집단에서 75%를 넘는 환자가 SF‑36의 물리적 구성요소에서 임상적으로 의미있는 악화 또는 사망을 경험하지 않았거나 (이 약 대 위약에 대해 75.1% 대 83.5%) 또는 SF‑36의 정신적 구성요소에서 임상적으로 의미있는 악화 또는 사망 (이 약 대 위약에 대해 77.7% 대 78.1%)을 경험하지 않았다. 이 약 군에서 SF‑36의 물리적 구성요소 또는 사망에 대한 약화 시간 (time to deterioration, TTD)이 짧은 경향이 관찰되었으며 (HR=1.43, 95% CI:0.96, 2.13), 두 군 모두에서 중앙값 TTD에 도달하지 않았다. SF‑36의 정신적 구성요소 또는 사망에 대한 TTD에서 군 간의 차이는 없었으며 (HR=0.90, 95% CI: 0.61, 1.33), 중앙값 TTD는 이 약 군에서 39.0개월 (95% CI: NC, NC)이고 위약 군에서는 도달하지 않았다.
절제 불가능한 국소 진행성(III기) EGFR 돌연변이 양성 비소세포폐암 – LAURA
확정적 백금‑기반 화학 방사선 요법 중 또는 후 진행되지 않은, EGFR 돌연변이 양성의 절제 불가능한 국소 진행성(III기) 비소세포폐암 환자의 치료에 대한 이 약의 유효성 및 안전성은 무작위 배정, 이중 눈가림, 위약 대조 시험(LAURA)에서 입증되었다. 환자들은 동시적 화학 방사선 치료(CCRT)나 순차적 화학 방사선 치료(SCRT) 요법을 받았으며, 여기에서 최소 2주기 또는 주 5 회 용량의 백금‑기반 화학 요법과 총 60 Gy ±10% (54 Gy ‑ 66 Gy) 용량의 방사선 치료가 무작위 배정 전 6주 이내에 완료되었다. 환자 종양 조직 검체는 공인/승인된 시험을 이용하여 중앙 또는 현지 검사로 확인된 EGFR 엑손 19 결손 또는 엑손 21 L858R 변이가 있어야 했다. 확정적 백금‑기반 화학 방사선 요법 중 또는 후 2등급 이상의 폐염증이 있거나 또는 화학 방사선 요법 이전에 간질성 폐염증의 병력이 있는 환자는 제외되었다.
환자들은 이 약 80 mg을 1일 1회 경구로(n=143) 또는 위약(n=73)을 투여받도록 무작위 배정되었다(2:1). 무작위 배정은 선행 화학 방사선 치료 전략(CCRT vs SCRT), 화학 방사선 치료 전 종양 병기 결정(IIIA vs IIIB/IIIC), 및 중국 코호트에 따라 층화되었다. 환자들은 치료에 대한 불내성 또는 확인된 질병 진행까지 시험 치료를 계속 받아야 했다. 이 후, 모든 환자는 치료 의사의 소견에 따라 기대되는 임상적 유익성이 있는 경우 현지 임상진료에 따라 라벨이 공개된 이 약을 제공받았다.
일차 유효성 평가변수는 눈가림된 독립적 중앙 검토(BICR)로 평가된 PFS였다. 추가 유효성 평가변수에는 CNS PFS, OS, ORR, DoR, 사망 또는 원격 전이까지 걸린 시간(TTDM), 첫 번째 후속 치료 시작 후 두 번째 PFS (PFS2), 첫 번째 후속 치료 또는 사망까지 걸린 시간(TFST), 및 두 번째 후속 치료 또는 사망까지 걸린 시간(TSST)이 포함되었다. CNS PFS, ORR, DoR, 및 TTDM은 모두 BICR로 평가되었다. 또한 환자 보고 결과도 평가되었다.
전체 시험대상군의 베이스라인 인구학적 및 질병 특성은 다음과 같았다: 연령 중앙값 63세(범위 36‑84세), ≧75세(13%), 여성(61%), 아시아인(82%), 백인(14%), 흡연력이 없는 사람(70%). 베이스라인 WHO 수행 능력 상태는 0 (51%) 또는 1 (49%)이었다; 환자의 35%는 IIIA기, 49%는 IIIB기, 그리고 16%는 IIIC기 비소세포폐암이었다. 환자의 조직학적 특성으로는 96%는 비편평세포암이었고, 4%는 편평세포암이었다. EGFR 돌연변이 상태와 관련해서는 54%가 엑손 19 결손이었고 45%가 엑손 21 L858R 변이였다. 무작위 배정 전에 환자의 89%는 CCRT를 받았고 11%는 SCRT를 받았다. 모든 환자는 백금‑기반 화학 요법(55%는 카보플라틴‑기반 화학 요법, 44%는 시스플라틴‑기반 화학 요법)을 받았다. 총 방사선량의 중앙값은 두 군에서 모두 환자에서 60 Gy였다.
백금‑기반 화학 방사선 요법 후 이 약의 치료는 위약 대비 PFS의 통계적으로 유의하고 임상적으로 유의미한 개선을 가져왔다(56% 성숙; HR=0.16; 95% CI: 0.10, 0.24; P<0.001, 각각 중앙값 39.1개월 및 5.6개월). 6, 12, 18, 24 및 36개월에서 이 약으로 치료 받은 환자의 무진행 생존 비율(각각 84%, 74%, 67%, 65% 및 58%)은 위약으로 치료받은 환자(각각 45%, 22%, 14%, 13% 및 10%)의 무진행 생존 비율보다 더 컸다.
임상시험 계획서에 따라, 모든 환자는 베이스라인 자기 공명 영상(MRI) 뇌 스캔을 받았고, 치료 중에 1명을 제외하고 모두 예정된 MRI 뇌 스캔을 받았다. 위약과 비교해 이 약으로 치료받은 환자에서 CNS PFS (신경방사선학자 BICR 평가에 근거)의 명목상 통계적으로 유의하고 임상적으로 유의미한 개선이 있었다(27% 성숙; HR=0.17; 95% CI: 0.09, 0.32; P<0.001 [명목]). 위약군에 비해 이 약 군에서 더 낮은 비율의 환자가 신경방사선학자 검토에 따라 새로운 CNS 병변이 있었다(각각 17/143 [12%] vs 26/73 [36%]).
전체 생존 기간의 중간 분석에서, 이 약에 유리한 긍정적 경향이 있었지만(20% 성숙; HR=0.81; 95% CI: 0.42, 1.56; P=0.530) 통계적으로 유의하지 않았다.
LAURA 임상시험에서 얻은 유효성 결과는 표 10에 요약되어 있으며, PFS와 CNS PFS에 대한 Kaplan‑Meier 곡선은 각각 그림 5 및 6에 나타내었다.
표 10. LAURA에서 얻은 유효성 결과
|
유효성 지표 |
이 약 (N=143) |
위약 (N=73) |
|
무진행 생존 기간 (Progression-Free Survival) | ||
|
사건 수(%) |
57 (40) |
63 (86) |
|
PFS 중앙값, 개월(95% CI) |
39.1 (31.5, NC) |
5.6 (3.7, 7.4) |
|
HR (95% CI); P-값 |
0.16 (0.10, 0.24); P<0.001 | |
|
CNS 무진행 생존 기간 (CNS Progression-Free Survival) | ||
|
사건 수(%) |
29 (20) |
30 (41) |
|
CNS PFS 중앙값, 개월(95% CI) |
NC (NC, NC) |
14.9 (7.4, NC) |
|
HR (95% CI); P-값 |
0.17 (0.09, 0.32); P<0.001 | |
|
12개월차 CNS 무진행 생존율, % (95% CI) |
87 (79, 92) |
53 (38, 66) |
|
24개월차 CNS 무진행 생존율, % (95% CI) |
83 (75, 89) |
43 (28, 58) |
|
전체 생존 기간 (Overall survival) | ||
|
사망 수(%) |
28 (20) |
15 (21) |
|
OS 중앙값, 개월(95% CI) |
54.0 (46.5, NC) |
NC (42.1, NC) |
|
HR (95% CI); P-값 |
0.81 (0.42, 1.56); P=0.530a | |
|
객관적 반응률b | ||
|
반응 수(n), 반응률 % (95% CI) |
82 57 (49, 66) |
24 33 (22, 45) |
|
오즈비(95% CI); P-값c |
2.77 (1.54, 5.08); P<0.001 | |
|
반응 기간(Duration of Response, DoR)b | ||
|
DoR 중앙값, 개월(95% CI)d |
36.9 (30.1, NC) |
6.5 (3.6, 8.3) |
|
사망 또는 원격 전이까지 걸린 시간(TTDM) | ||
|
환자 수(%) |
33 (23) |
31 (43) |
|
TTDM 중앙값, 개월(95% CI) |
NC (39.3, NC) |
13.0 (9.0, NC) |
|
HR (95% CI); P-값 |
0.21 (0.11, 0.38); P<0.001 | |
|
첫 번째 후속 치료 시작 후 두 번째 PFS (PFS2) | ||
|
PFS2 사례 수(%) |
34 (24) |
24 (33) |
|
PFS2 중앙값, 개월(95% CI) |
48.2 (44.4, NC) |
47.4 (28.2, NC) |
|
HR (95% CI); P-값 |
0.62 (0.35, 1.08); P=0.088 | |
|
무작위 배정부터 첫 번째 후속 치료 또는 사망까지의 시간(TFST) | ||
|
첫 번째 후속 치료를 받거나 사망한 환자 수(%) |
53 (37) |
61 (84) |
|
TFST 중앙값, 개월(95% CI) |
43.8 (38.9, NC) |
9.5 (6.6, 11.5) |
|
HR (95% CI); P-값 |
0.13 (0.08, 0.21); P<0.001 | |
|
무작위 배정부터 두 번째 후속 치료 또는 사망까지의 시간(TSST) | ||
|
두 번째 후속 치료를 받거나 사망한 환자 수(%) |
32 (22) |
24 (33) |
|
TSST 중앙값, 개월(95% CI) |
NC (44.4, NC) |
47.4 (34.1, NC) |
|
HR (95% CI); P-값 |
0.51 (0.28, 0.91); P=0.022 | |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, CNS PFS, ORR, DoR, 및 TTDM 결과는 BICR에 따라 평가하였다.
모든 환자에서 PFS에 대한 추적관찰 시간 중앙값은 이 약 군에서 22.0개월, 위약군에서 5.6개월이었다.
HR <1 및 오즈비 >1은 이 약에 유리하다.
a 중간 분석(20% 성숙)에 대해 보정한 결과 통계적 유의성에 도달하기 위해서는 p 값<0.00036이 필요했다.
b 확인되지 않은 응답을 기반으로 했다.
c 분석은 화학 요법 전 병기(IIIA vs IIIB/IIIC)에 따라 계층화된 로지스틱 회귀 분석을 사용하여 수행되었다.
d Kaplan‑Meier 방법으로 계산했다.
그림 5. LAURA에서 BICR로 평가된 PFS에 대한 Kaplan‑Meier 곡선
위약 대비 이 약의 PFS 유익성은 성별, 무작위 배정 시점의 연령, 흡연력, 민족성, 선행 화학 방사선 치료 전략(CCRT vs SCRT), 화학 방사선 치료 전 병기(IIIA vs IIIB/IIIC), 선행 화학 방사선 치료에 대한 반응, 및 EGFR 돌연변이 유형(엑손 19 결손 또는 L858R)을 포함하여 분석된 사전 정의된 모든 하위군에서 일관적이었다.
그림 6. LAURA에서 BICR로 평가된 CNS PFS의 Kaplan‑Meier 곡선
이 약에 무작위 배정된 환자는 위약에 무작위 배정된 환자에 비해 PFS2, TFST 및 TSST에서 임상적으로 유의미한 개선이 있었다. 이러한 진행 후 평가변수의 분석은 이 약으로 교차 투여 수준이 높음에도 불구하고 PFS 유익성이 후속 차수 치료를 통해 유지됨을 입증하였다.
환자 보고 결과
환자 보고 증상 및 HRQL 자료는 EORTC QLQ‑C30 (C30) 및 폐암 모듈 EORTC QLQ‑LC13 (LC13)을 이용하여 전자적으로 수집되었다. 베이스라인에서 환자 보고 증상, 신체적 기능 및 전반적인 건강 상태/삶의 질(GHS/QoL)은 이 약 군과 위약군 간에 유사하였다. 환자가 보고한 5가지 주요 폐암 및 치료 관련 증상(기침, 호흡 곤란, 흉통, 피로, 및 식욕 상실)과 신체적 기능 영역 및 GHS/QoL의 베이스라인 대비 전체 평균 변화에서 임상적으로 유의미한 차이는 없었다. 치료군 간에 이러한 환자 보고 증상, 신체적 기능 또는 GHS/QoL의 악화 위험에서 유의미한 차이는 관찰되지 않았다.
이전에 치료 받은 적 없는 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 – FLAURA –단독요법
진행성 질환에 대하여 이전에 전신 치료를 받지 않은 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 치료 시 이 약의 유효성 및 안전성이 무작위배정, 이중 눈가림, 활성 대조 시험(FLAURA)에서 입증되었다.
환자들은 이 약 (n=279, 80 mg 1일 1회 경구 투여) 또는 EGFR TKI 대조약(n=277; 게피티닙 250 mg 1일 1회 경구 투여 또는 엘로티닙 150 mg 1일 1회 경구 투여)을 투여 받도록 1:1로 무작위 배정 되었다. 무작위 배정은 EGFR 변이 유형(Ex19del 또는 L858R) 및 민족성(아시아인 또는 비아시아인)에 따라 층화되었다. 환자들은 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더 이상 임상적 유익성을 경험하지 않을 때까지 시험 치료를 투여 받았다. EGFR TKI 대조약을 투여 받은 환자의 경우, 진행이 발생한 후 종양 샘플 검사 시 T790M 변이에 양성인 경우 이 약 공개라벨으로 교차가 허용되었다.
이 약은 EGFR TKI 대조약과 비교하여 임상적으로 의미 있고 통계적으로 크게 유의한 PFS 개선을 입증하였다 (중앙값 각각 18.9 개월 및 10.2 개월, HR=0.46, 95% CI: 0.37, 0.57; P<0.0001). 연구자 평가에 의한 FLAURA에서의 유효성 결과가 표 11에 요약되어 있으며, PFS에 대한 Kaplan‑Meier 곡선을 그림 7에 나타내었다. 전체 생존 기간 최종 분석 (58% 성숙)은 EGFR TKI 대조약과 비교하여 이 약에 무작위 배정된 환자에서 0.799의 HR (95% CI: 0.641, 0.996; P = 0.0462)로 통계적으로 유의한 개선 및 임상적으로 의미 있는 보다 긴 중앙 생존 기간을 입증했다 (표 11 및 그림 8). 12개월, 18개월, 24개월 및 36개월 시점에 생존한 환자 비율은 이 약 (각각 89%, 81%, 74% 및 54%) 치료 시 EGFR TKI 대조약 (각각 83%, 71%, 59% 및 44%) 치료에 비해 높았다.
표 11. 연구자 평가에 따른 FLAURA에서의 유효성 결과
|
유효성 지표 |
이 약 (N=279) |
EGFR TKI 대조약 (게피티닙 또는 엘로티닙) (N=277) |
|
무진행 생존 기간(Progression-Free Survival) | ||
|
사건 수 (62% 성숙) |
136 (49) |
206 (74) |
|
PFS 중앙값, 개월 (95% CI) |
18.9 (15.2, 21.4) |
10.2 (9.6, 11.1) |
|
HR (95% CI); P-값 |
0.46 (0.37, 0.57); P<0.0001 | |
|
전체 생존 기간(Overall Survival) | ||
|
사망 수, (58% 성숙) |
155 (56) |
166 (60) |
|
OS 중앙값, 개월 (95% CI) |
38.6 (34.5, 41.8) |
31.8 (26.6, 36.0) |
|
HR (95% CI); P-값 |
0.799 (0.641, 0.997); P=0.0462a | |
|
객관적 반응률(Objective Response Rate)*b | ||
|
반응자 수 (n), 반응률 % (95% CI) |
223 80 (75, 85) |
210 76 (70, 81) |
|
오즈비 (95% CI); P-값 |
1.3 (0.9, 1.9); P=0.2421 | |
|
반응 기간 (Duration of Response, DoR)* | ||
|
DoR 중앙값, 개월 (95% CI) |
17.2 (13.8, 22.0) |
8.5 (7.3, 9.8) |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, ORR, DoR 결과는 RECIST 연구자 평가에 근거한 것이다.
*확인되지 않은 반응에 근거한 것이다.
PFS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 15.0개월이었고 EGFR TKI 대조약을 투여 받은 환자에서 9.7개월이었다.
OS 추적관찰 시간의 중앙값은 이 약을 투여 받은 환자에서 35.8개월이었고 EGFR TKI 대조약을 투여 받은 환자에서 27.0개월이었다.
PFS, ORR, DoR 결과는 자료마감일 (data cut‑off) 2017년 6월 12일이다. OS 결과는 자료마감일 2019년 6월 25일이다.
HR <1 및 오즈비 >1은 이 약에 유리하다.
a 중간 분석(25% 성숙)을 위해 조정된 통계적 유의성을 달성하려면 p‑값 <0.0495이 필요했다
b 눈가림된 독립적 중앙 검토(BICR)에서 평가한 ORR 결과는 연구자 평가에서 보고된 것과 일치하였다; BICR 평가에 따른 ORR은 이 약에서 78% (95% CI: 73, 83)였고 EGFR TKI 대조약에서 70% (95% CI: 65, 76)였다.
그림 7. FLAURA에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
FLAURA2 – 병용요법
진행성 질환에 대하여 이전에 전신 치료를 받지 않은 EGFR 변이 양성 국소 진행성 또는 전이성 비소세포폐암 환자 치료 시 페메트렉시드와 백금 기반 항암화학요법과 병용하는 이 약의 유효성 및 안전성이 무작위배정, 공개, 활성 대조 시험 (FLAURA2)에서 입증되었다. 환자의 종양 조직 샘플은 현지 또는 중앙 검사로 확인된, EGFR TKI 민감도와 관련이 있는 것으로 알려진 두 가지 일반적인 EGFR 변이 (Ex19del 또는 L858R) 중 하나가 있어야 했다.
환자들은 다음의 치료군 중 하나에 무작위 배정되었다 (1:1):
‧ 이 약 (80 mg) 1일 1회 경구 투여 시, 페메트렉시드 (500 mg/m2) 그리고 시스플라틴 (75 mg/m2) 또는 카보플라틴 (AUC5) 중 연구자가 선택한 약물을 4주기 동안 21일 주기의 Day1에 정맥 내 투여한 후, 이 약 (80 mg) 1일 1회 경구 투여 및 페메트렉시드 (500 mg/m2) 3주마다 정맥 내 투여 (n=279)
‧ 이 약 (80 mg) 1일 1회 경구 투여 (n=278)
무작위 배정은 인종 (중국인/아시아인, 비중국인/아시아인 또는 비아시아인), WHO 수행 능력 (0 또는 1), 및 조직 검사 방법 (중앙 또는 현지)에 따라 층화되었다. 환자는 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더 이상 임상적 유익성을 경험하지 않을 때까지 시험 치료를 투여 받았다.
일차 유효성 평가변수는 RECIST 1.1에 따라 연구자가 평가한 PFS였다. 추가적인 유효성 평가변수에는 연구자의 평가에 따른 OS, ORR, DoR, PFS2, TFST 및 TSST가 포함되었다. BICR에 의해 평가된 CNS PFS, CNS ORR, CNS DoR 그리고 PRO도 평가되었다.
전체 시험 집단에 대한 베이스라인 인구통계학적 특성 및 질병 특성은 다음과 같았다: 연령 중앙값 61세 (범위 26‑85세), ≧75세 (8%), 여성 (61%), 아시아인 (64%), 백인 (28%), 흡연 미경험자 (66%). 모든 환자는 WHO 수행 능력이 0 또는 1이었다; 환자의 49%는 전이성 골 질환이 있었고, 환자의 53%는 흉곽 외 전이가 있었으며 20%는 간 전이가 있었다. 환자의 41%는 CNS 전이가 있었다 (베이스라인에 CNS 병변 부위, CNS 전이에 대한 병력, 및/또는 이전 수술, 및/또는 이전 방사선요법을 근거로 연구자가 확인함).
페메트렉시드 및 백금 기반 항암화학요법과 병용하는 이 약은 이 약의 단일요법과 비교하여 임상적으로 의미 있고 통계적으로 유의한 PFS 개선을 입증하였다 (51% 성숙; HR=0.62, 95% CI: 0.49, 0.79; P<0.0001; 중앙값 각각 25.5개월 및 16.7개월). OS 중간 분석 시점에, 페메트렉시드 및 백금 기반 항암화학요법과 병용한 이 약 투여 군의 환자들은 이 약의 단일요법 군에 비해 OS에 영향을 받지 않았다 (27% 성숙; HR=0.90, 95% CI: 0.65, 1.24; P = 0.5238).
연구자 평가에 따른 FLAURA2의 유효성 결과는 표 12에 요약되어 있으며, PFS에 대한 Kaplan‑Meier 곡선은 그림 9에 나타내었다.
표 12. 연구자 평가에 따른 FLAURA2에서의 유효성 결과
|
유효성 지표 |
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=279) |
이 약 (N=278) |
|
무진행 생존 기간(Progression-Free Survival) | ||
|
사건 수 (%) |
120 (43) |
166 (60) |
|
PFS 중앙값, 개월 (95% CI) |
25.5 (24.7, NC) |
16.7 (14.1, 21.3) |
|
HR (95% CI); P-값 |
0.62 (0.49, 0.79); P<0.0001 | |
|
전체 생존 기간(Overall Survival) | ||
|
사망 수 (%) |
71 (25) |
78 (28) |
|
OS 중앙값, 개월 (95% CI) |
NC (31.9, NC) |
NC (NC, NC) |
|
HR (95% CI); P-값 |
0.90 (0.65, 1.24); P=0.5238a | |
|
객관적 반응률(Objective Response Rate)b,c | ||
|
반응자 수 (n), 반응률 % (95% CI) |
232 83 (78, 87) |
210 76 (70, 80) |
|
오즈비 (95% CI); P-값d |
1.61 (1.06, 2.44); P=0.0261 | |
|
반응 기간 (Duration of Response, DoR)b | ||
|
DoR 중앙값, 개월 (95% CI)e |
24.0 (20.9, 27.8) |
15.3 (12.7, 19.4) |
|
질병 조절률 (Disease Control Rate) | ||
|
질병 조절되는 환자 수 (n), 조절률 % (95% CI) |
266 95 (92, 98) |
261 94 (90, 96) |
|
오즈비 (95% CI); P-값d |
1.33 (0.63, 2.81); P=0.4483 | |
|
첫 번째 후속 치료 시작 후 두 번째 PFS(PFS2) | ||
|
PFS2 사건 수 (%) |
81 (29) |
110 (40) |
|
PFS2 중앙값, 개월 (95% CI) |
30.6 (29.0, NC) |
27.8 (26.0, NC) |
|
HR (95% CI); P-값 |
0.70 (0.52, 0.93); P=0.0132 | |
|
무작위 배정부터 첫 번째 후속 치료 또는 사망까지의 시간(TFST) | ||
|
첫 번째 후속 치료를 받았거나 사망한 환자 수(%) |
104 (37) |
129 (46) |
|
TFST 중앙값, 개월 (95% CI) |
30.7 (27.3, NC) |
25.4 (22.8, NC) |
|
HR (95% CI); P-값 |
0.73 (0.56, 0.94); P=0.0159 | |
|
무작위 배정부터 두 번째 후속 치료 또는 사망까지의 시간(TSST) | ||
|
두 번째 후속 치료를 받았거나 사망한 환자 수(%) |
74 (27) |
103 (37) |
|
TSST 중앙값, 개월 (95% CI) |
NC (NC, NC) |
33.2 (28.2, NC) |
|
HR (95% CI); P-값 |
0.69 (0.51, 0.93); P=0.0157 | |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
PFS, ORR, DoR, DCR 결과는 RECIST 연구자 평가에 근거한 것이다.
PFS 추적관찰 시간의 중앙값은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 받은 환자에서 19.5개월이었고 이 약의 단독요법 군에서는 16.5개월이었다.
HR <1 및 오즈비 >1은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에 유리하다.
a 중간 분석(27% 성숙)에 따르면 통계적 유의성을 달성하려면 p‑값 < 0.00158 이 필요했다.
b 확인되지 않은 응답을 기반으로 했다.
c BICR에서 평가한 ORR 결과는 연구자 평가에서 보고된 것과 일치하였다; BICR 평가에 따른 ORR은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에서 92% (95% CI: 88, 95)였고 이 약의 단독요법 군에서 83% (95% CI: 78, 87)였다.
d 분석은 인종 (중국인/아시아인 vs. 비중국인/아시아인 vs. 비아시아인), WHO 수행 능력 (0 또는 1), 및 조직 검사 방법 (중앙 또는 현지)에 따라 계층화된 로지스틱 회귀 분석을 사용하여 수행되었다.
e Kaplan‑Meier 방법으로 계산했다.
그림 9. FLAURA2에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
이 약의 단일요법 대비 페메트렉시드 및 백금 기반 항암화학요법과 병용하는 이 약의 PFS 유익성은 민족성, 연령, 성별, 흡연 유무, 시험 시작 시 CNS 전이 상태, 및 EGFR 돌연변이 유형 (엑손 19 결손 또는 L858R)을 포함하여 사전에 정의된 모든 분석 하위군에 걸쳐 일관성 있게 관찰되었다.
베이스라인 대비 표적 병변 크기의 가장 우수한 백분율 변화 중앙값은 페메트렉시드 및 백금 기반 항암화학요법과 이 약 병용 투여 군에서 ‑53% (범위: 100%~20%)였고 이 약의 단일요법 군에서는 ‑50% (범위: ‑100%~40%)였다.
1차 치료로서 페메트렉시드 및 백금 기반 항암화학요법과 이 약 병용 투여 군에 무작위 배정된 환자도 이 약의 단일요법 군에 무작위 배정된 환자에 비해 임상적으로 의미있는 PFS2, TFST 및 TSST 개선을 보였다. 이러한 진행 후 평가변수 분석은 PFS 유익성이 후속 차수의 치료까지 대부분 유지되었음을 입증하였다.
FLAURA2에서 CNS 전이 유효성 데이터
스테로이드가 필요하지 않은 무증상 CNS 전이 환자와 최종 치료 및 스테로이드 치료 완료 후 최소 2주 동안 신경학적 상태가 안정적인 환자는 FLAURA2 연구에 무작위 배정될 자격이 있었다. 모든 환자는 베이스라인 뇌 스캔이 가능했다. 수정된 RECIST를 사용한 BICR 평가에서는 CNS 측정 가능 및/또는 측정 불가능 병변(cFAS)이 있는 환자 222/557명(40%)의 하위 그룹과 CNS 측정 가능 병변(cEFR)이 있는 환자 78/557 (14%) 의 하위 그룹이 추가로 생성되었다. FLAURA2에 대한 BICR의 CNS 효능 평가 관련하여, 이 약의 단독요법 군에 비해 페메트렉시드와 백금 기반 항암화학요법과 이 약의 병용 투여 군에서 cFAS 및 cEFR에 대한 CNS PFS에서 임상적으로 의미 있는 개선을 입증했다(cFAS: 27% 성숙도, HR= 0.58, 95% CI 0.33, 1.01, P=0.0548[명목] 및 cEFR: 37% 성숙도, HR=0.40, 95% CI 0.19, 0.84, P=0.0157[명목]).
CNS 효능 데이터는 표 13에 요약되었다.
표 13. FLAURA2에서 베이스라인 뇌 스캔에서 CNS 전이가 있는 환자에 대해 BICR 평가된 CNS 유효성
|
유효성 지표 |
CNS 측정가능 및/또는 측정 불가능 병변 (cFAS) |
CNS 측정가능 병변 (cEFR) | ||
|
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=118) |
이 약 (N=104) |
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=40) |
이 약 (N=38) | |
|
CNS 무진행 생존 기간(CNS PFS)a | ||||
|
사건 수 (%) |
28 (24) |
31 (30) |
11 (28) |
18 (47) |
|
CNS PFS 중앙값, 개월 (95% CI) |
30.2 (28.4, NC) |
27.6 (22.1, NC) |
NC (23.0, NC) |
17.3 (13.9, NC) |
|
HR (95% CI); P-값 |
0.58 (0.33, 1.01); P=0.0548 |
0.40 (0.19, 0.84); P=0.0157 | ||
|
CNS 진행 없이 12개월 째 생존 (%) (95% CI) |
87 (79, 92) |
83 (73, 89) |
89 (74, 96) |
73 (55, 85) |
|
CNS 진행 없이 24개월 째 생존 (%) (95% CI) |
74 (63, 82) |
54 (39, 67) |
65 (43, 80) |
37 (18, 57) |
|
CNS 객관적 반응률a | ||||
|
CNS 반응 수(n), CNS 반응률 % (95% CI) |
86 73 (64, 81) |
72 69 (59, 78) |
35 88 (73, 96) |
33 87 (72, 96) |
|
완전 관해 (Complete response) |
70 (59) |
45 (43) |
19 (48) |
6 (16) |
|
부분 관해 (Partial response) |
16 (14) |
27 (26) |
16 (40) |
27 (71) |
|
오즈비 (95% CI); P-값b,c |
1.19 (0.67, 2.14); P=0.5492 |
1.06 (0.28, 4.00); P=0.9308 | ||
|
CNS Duration of Responsea | ||||
|
CNS DoR 중앙값, 개월 (95% CI) |
NC (23.8, NC) |
26.2 (19.4, NC) |
NC (21.6, NC) |
20.9 (12.6, NC) |
|
12개월 시점에 반응 보이는 환자 (%) (95% CI) |
93 (85, 97) |
81 (68, 89) |
93 (75, 98) |
74 (53, 87) |
|
24개월 시점에 반응 보이는 환자 (%) (95% CI) |
62 (40, 77) |
57 (38, 72) |
57 (27, 78) |
45 (22, 65) |
HR (Hazard Ratio) = 위험비; CI (Confidence Interval) = 신뢰 구간, NC (Not Calculable) = 계산 불가능
HR <1 및 오즈비 >1은 이 약과 페멕트렉시드 및 백금 기반 항암화학요법을 병용 투여 군에 유리하다.
a 확인되지 않은 응답을 기반으로 했다.
b 명목(Nominal) P‑값
c 분석은 치료 요인을에 대한 로지스틱 회귀분석을 이용하여 수행했다.
베이스라인 대비 표적 CNS 병변 크기의 최고 백분율 변화 중앙값은 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서 ‑94%(범위: ‑100% ~ 7%)였고, 이 약의 단독요법 군에서 ‑61%(범위: ‑100% ~ 68%)이었다.
FLAURA2에서는 CNS 전이 상태(기준시 CNS 병변 부위, 병력 및/또는 이전 수술 및/또는 CNS 전이에 대한 이전 방사선 치료를 기반으로 연구자가 식별함)를 기반으로 사전 지정된 PFS 하위군으로 연구를 시작했다. 연구 시작 시 CNS 전이 상태와 관계없이, 이 약의 단독요법 군에 비해 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서의 PFS 개선이 입증되었다. 연구 시작 시 CNS 전이 상태별 PFS에 대한 Kaplan‑Meier 곡선을 그림 10에 제시했다.
그림 10. FLAURA2에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
환자 보고 증상 및 HRQL은 EORTC QLQ C30 및 그에 대한 폐암 모듈 (EORTC QLQ‑LC13)을 이용해 전자적으로 수집되었다. LC13은 첫 8주 동안 주1회 시행되었고, 이후 진행 전에는 3주마다 그리고 진행 후에는 8주마다 시행되었다. C30은 첫 8주 동안 3주마다 평가되었고, 이후 진행 전에는 6주마다 그리고 진행 후에는 8주마다 평가되었다. 베이스라인에, 환자 보고 증상, 신체 기능 또는 GHS/QoL에서 페메트렉시드 및 백금기반 화학요법과 병용하는 이 약 군과 이 약의 단일요법군 간에 차이는 관찰되지 않았다. 첫 19개월에 걸친 순응도는 전반적으로 높았고 (≧80%) 두 군 모두 유사했다.
주요 폐암 증상 분석
베이스라인부터 제19개월까지 수집된 자료는 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군과 이 약의 단일요법 군에서 사전 명시된 5가지 일차 PRO 증상 중 3가지 (기침, 호흡곤란, 및 흉통)에 대해 유사한 개선을 나타냈으며, 기침에서의 개선은 두 군 모두 확립된 임상적으로 관련된 기준치 (베이스라인 대비 변화 ≦‑10)에 도달했다. 이 약의 단일요법군에서는 식욕 상실 및 피로에 대해 개선되는 추세를 보였다. 페메트렉시드 및 백금 기반 항암화학요법과 이 약의 병용 투여 군에서는 화학요법의 첫 4주기 동안 피로가 악화되는 추세를 보이다가 이후 개선되는 추세를 보였으며, 식욕 상실은 악화되는 추세를 보였다. 이러한 변화는 임상적으로 의미가 없었다. 이러한 자료는 표 14에 제시되어 있다.
표 14. 혼합 모델 반복 측정 ‑ 주요 폐암 증상 – 이 약 단독 요법으로 치료받은 환자와 페메트렉시드 및 백금 기반 항암화학요법과 이 약을 병용하여 치료받은 환자의 베이스라인으로부터의 평균 변화
|
|
이 약과 페멕트렉시드와 백금 기반 항암화학요법 병용 (N=279) |
이 약 (N=278) |
치료에서의 예상되는 차이 (95%CI) | ||
|
N |
Adjusted mean |
N |
Adjusted mean | ||
|
기침 |
253 |
-13.23 |
251 |
-11.19 |
-2.04 (-4.35, 0.26) |
|
호흡 곤란 |
253 |
-3.09 |
251 |
-5.67 |
2.57 (0.28, 4.86) |
|
가슴 통증 |
253 |
-6.33 |
251 |
-6.61 |
0.29 (-1.62, 2.20) |
|
식욕 감퇴 |
253 |
2.87 |
253 |
-4.58 |
7.45 (4.52, 10.38) |
|
피로감 |
253 |
-0.03 |
253 |
-6.31 |
6.28 (3.60, 8.96) |
전반적인 건강상태/QoL 및 신체기능 개선 분석
두 군 모두 신체 기능 및 GHS/QoL에 임상적으로 의미 있는 변화가 없다고 보고했다.
이전에 치료 받은 적 있는 T790M양성 비소세포폐암 환자 ‑ AURA3
TKI 치료 후 또는 치료 중 진행되면서 국소진행 또는 전이된 T790M 비소세포폐암 환자 치료 시 이 약의 유효성 및 안전성은 무작위 배정된, 공개, 활성 대조군 임상 3상시험(AURA 3)에서 증명되었다.
환자들은 이 약 (n=279) 또는 백금기반 이중 항암화학요법제(n=140)에 2:1 (이 약: 백금기반 이중 항암화학요법제) 비율로 무작위 배정되었다. 무작위 배정은 민족성 (아시아인 및 비아시아인)에 따라 층화되었다. 이 약 군의 환자는 치료에 불내성이 생길 때까지 또는 연구자가 판단하기에 환자가 더이상 임상적 유익성을 경험하지 않을 때까지 이 약 80 mg을 1일 1회 경구 투여 받았다.
화학요법은 최대 6주기동안 매 21일 주기의 제 1일째에 페메트렉시드 500mg/m2와 카보플라틴 AUC5 병용요법 또는 페메트렉시드 500mg/m2와 시스플라틴 75mg/m2병용요법으로 이루어졌다. 4주기의 백금기반 화학요법 후에 질병이 진행되지 않은 환자는 페메트렉시드 유지요법을 받을 수 있었다 (매 21일 주기의 제 1일째에 페메트렉시드 500mg/m2). 화학요법군의 시험대상자 중 객관적인 방사선학적 진행이 있는 환자 (연구자에 의해 그리고 독립적인 중앙 영상 검토에 의해 확정된)는 이 약으로 치료를 시작할 수 있는 기회가 주어졌다.
AURA 3시험에서 항암화학요법군 대비 이 약으로 치료받은 환자군에서 통계적으로 유의한 PFS 개선이 증명되었다. AURA 3 연구자 평가에 따른 AURA 3의 유효성 결과는 표 15에 요약되어 있고, 그림 11에 PFS에 대한 Kaplan‑Meier 곡선을 나타내었다. 최종 OS 분석 (67 % 성숙 시 실시)에서 치료군 간에 통계적으로 유의한 차이가 관찰되지 않았으며, 이 시점에는 항암화학요법에 무작위 배정된 99명의 환자 (71 %)가 이 약 치료로 넘어가 있었다.
표 15. 연구자 평가에 따른 AURA3에서의 유효성 결과
|
유효성 지표 |
이 약(N=279) |
항암화학요법 (N=140) |
|
무진행생존기간(Progression-Free Survival) | ||
|
사건 수 (% maturity) |
140 (50) |
110 (79) |
|
PFS 중앙값, 개월 (95% CI) |
10.1 (8.3, 12.3) |
4.4 (4.2, 5.6) |
|
HR (95% CI) ; P-값 |
0.30 (0.23,0.41); P <0.001 | |
|
전체 생존기간(Overall Survival, OS)1 | ||
|
사망 수 (% maturity) |
188 (67.4) |
93 (66.4) |
|
OS 중앙값, 개월 (95% CI) |
26.8 (23.5, 31.5) |
22.5 (20.2, 28.8) |
|
HR (95.56% CI); P-값 |
0.87 (0.67, 1.13); P=0.277 | |
|
객관적 반응률(Objective Response Rate)2 | ||
|
반응자 수, 반응률 (95% CI) |
197 71% (65, 76) |
44 31% (24, 40) |
|
오즈비 (95% CI) ; P-값 |
5.4 (3.5, 8.5); P <0.001 | |
|
반응 기간(Duration of Response, DoR)2 | ||
|
DoR 중앙값, 개월 (95% CI) |
9.7 (8.3, 11.6) |
4.1 (3.0, 5.6) |
HR=위험비; CI=신뢰구간; OS=전체 생존기간
RECIST 연구자 평가에 근거한 모든 유효성 결과
HR <1 및 오즈비 >1은 이 약에 유리하다.
1 OS의 최종 분석은 67% 성숙 시 실시되었다. HR에 대한 CI는 이전 중간 분석을 위해 조정되었다. OS 분석은 교차에 대한 잠재적인 교란혼재효과는 보정되지 않았다(항암화학요법군의 99명 [71%] 환자는 이후 오시머티닙 치료를 받았다.).
2 연구자 평가에 의한 ORR 및 DoR 결과는 눈가림된 독립적 중앙 검토(Blinded Independent Central Review, BICR)를 통해 보고된 것과 일치한다; BICR 평가에 의한 ORR은 오시머티닙 치료 시 64.9% [95% CI: 59.0, 70.5], 항암화학요법제 치료 시 34.3% [95% CI: 26.5, 42.8]였다. ; BICR에 의한 DoR은 오시머티닙 치료 시 11.2개월(95% CI: 8.3, NC), 항암화학요법제 치료 시 3.1 개월(95% CI: 2.9, 4.3)이었다.
그림 11. AURA3에서 연구자에 의해 평가된 PFS에 대한 Kaplan‑Meier 곡선
항암화학요법제를 투여 받은 환자군과 비교하여 이 약을 투여 받은 환자군에게 유리한 0.50 미만 HR을 보이는 임상적으로 의미 있는 PFS 개선은 민족성, 나이, 성별, 흡연 유무, 시험 시작 시 CNS 전이 상태, EGFR 변이 (엑손 19 결손 또는 L858R), 및 EGFR‑TKI 1차 치료 기간을 포함하여 사전에 정의된 모든 분석 하위군에서 일관성 있게 관찰되었다.
4) 독성시험 정보
반복 투여 독성
랫드와 개에 대한 반복 투여 독성시험에서 관찰된 주요 결과는 눈(각막), 위장관(혀 포함), 피부 및 수컷과 암컷 생식기관의 상피에 영향을 미치는 위축성, 염증성 그리고/또는 퇴행성 변화로 구성되었다. 이러한 결과는 80 mg 치료 용량의 환자들에서 관찰된 것보다 낮은 혈장 농도에서 발생하였다. 투여 1개월 후 발생한 결과는 투여 중단 후 1개월 이내에 대체로 회복을 나타내었다.
발암성 및 돌연변이성
오시머티닙은 26주 동안 Tg rasH2 유전자 이식 마우스에게 경구 투여했을 때 발암 가능성을 보이지 않았다.
랫드의 104주 발암성 연구에서 1일 1회 80 mg의 권장 임상 용량에서 관찰된 AUC의 0.2배 노출에서 수정체 섬유 변성 및 장간막 림프절에서 증식성 혈관 병변 (혈관종 과형성 및 혈관종)의 발생률 증가가 관찰되었다. 수정체 섬유 변성은 52주차에 처음 관찰되고 투여 기간이 증가함에 따라 발생률 및 중증도가 점진적으로 증가하는 수정체 혼탁에 대한 검안경 관찰과 일치했다. 이러한 결과의 임상적 연관성은 배제할 수 없다. 장간막 림프절에서의 증식성 혈관 병변(혈관종 과형성 및 혈관종)은 인간의 혈관 신생물에 대한 발암 가능성을 예측하는 것은 아니다.
오시머티닙은 in vitro 및 in vivo 시험에서 유전적 손상을 유발하지 않았다.
생식 독성
동물에 대한 시험에 근거하여 수컷 수태능은 이 약 치료로 손상될 수 있다. 1개월 이상 오시머티닙에 노출된 랫드와 개에서 퇴행성 변화를 고환에서 보였으며, 3개월 동안 오시머티닙에 노출된 후 랫드에서 수컷 수태능이 감소하였다. 이러한 결과들은 임상적으로 관련된 혈장 농도에서 관찰되었다. 1개월 투여 후 보인 고환에서의 병리학적 소견은 랫드에서 가역적이었으나, 개에서 이러한 병변의 가역성에 대한 확정적인 진술은 할 수 없다.
동물에서의 시험에 근거하여 암컷 수태능은 이 약 치료로 손상될 수 있다. 반복투여 독성시험에서 임상적으로 관련된 혈장 농도로 오시머티닙에 1개월 이상 노출된 랫드에서 발정휴지기 증가, 난소에서 황체 퇴행 및 자궁과 질의 상피 얇아짐이 관찰되었다. 1개월 투여 후 관찰된 난소에서의 결과는 가역적이었다. 랫드에 대한 암컷 수태능 시험에서, 오시머티닙 20 mg/kg/day 투여 (권장 1일 임상 용량인 80 mg과 거의 동등한 용량)는 발정 주기 또는 임신하는 암컷의 수에 영향이 없었으나, 초기 배아 사망을 야기했다. 이러한 결과는 1개월간 휴약 후 가역성의 증거를 보였다.
랫드에 대한 수정된 배태자 발달 시험에서, 오시머티닙은 임신한 랫드에게 배아 착상 전 투여 시 배아 치사를 초래했다. 이러한 영향은 권장 용량 1일 80 mg에서의 사람 노출과 동등한 노출 (총 AUC에 근거)인 20 mg/kg/day의 모체 내약 용량 (maternally tolerated dose)에서 관찰되었다. 기관 형성기에 20 mg/kg 이상의 용량에 대한 노출은 태자 무게 감소를 유발하였으나, 태자의 외형 또는 내장 형태에는 유해한 영향을 미치지 않았다. 오시머티닙을 임신한 암컷 랫드에게 임신 기간에 걸쳐서, 그리고 이후 수유 초기에 투여했을 때, 수유를 받는 차산자에서 오시머티닙 및 그 대사체에 대한 노출을 보였으며, 차산자의 생존 감소 및 차산자 성장 감소가 있었다. (20 mg/kg 이상의 용량에서)