
| 제품명 |
|
|||
|---|---|---|---|---|
| 성분 / 함량 |
|
|||
| 첨가제 | ||||
| 도핑금지 약물정보 |
|
|||
| 전문 / 일반 |
|
단일 / 복합 | ||
| 제조 / 수입사 | ||||
| 제형 | 투여경로 | |||
| 성상 | ||||
| 허가일 |
|
|||
| 재심사 | ||||
| 대조 / 생동 | ||||
| 급여정보 |
624900101 - 삭제(2021-11-01) - 277123원/0.8mL/펜 급여(2021-06-07) - 395890원/0.8mL/펜 급여(2019-09-01) - 406458원/0.8mL/펜 급여(2018-09-01) - 414753원/0.8mL/펜 급여(2018-02-01) - 414850원/0.8mL/펜 급여(2017-02-01)
|
|||
| ATC 코드 | ||||
| KPIC 약효분류 |
|
|||
| KPIC 학술 |
팜리뷰
건선치료의 최신동향, 약학정보원(정다솜 약사), 2026-01-12
팜리뷰
[Trend Focus] 류마티스 관절염 치료제의 최신 동향, 약학정보원(김수연), 2022-04-15
팜리뷰
[Drug Safety Report] 항류마티스 약물(DMARDs)의 안전성 정보와 사례연구, 약학정보원(대한약사회 지역의약품안전센터), 2022-04-08
팜리뷰
[Pharmacotherapy Today] 류마티스 관절염 치료의 약물요법, 약학정보원(부산대학교병원 약제부 최은경), 2022-04-01
팜리뷰
류마티스 관절염의 약물요법, 약학정보원(송영천), 2018-04-30
팜리뷰
건선, 어떻게 대할 것인가(2), 약학정보원(최혁재), 2018-03-19
팜리뷰
건선, 어떻게 대할 것인가(1), 약학정보원(최혁재), 2018-03-12
팜리뷰
크론병(2), 약학정보원(최혁재), 2016-09-26
팜리뷰
크론병(1), 약학정보원(최혁재), 2016-09-19
팜리뷰
특이적 약인성 간손상, 약학정보원(최선), 2013-12-16
팜리뷰
[안전] 혈관에 영향을 미치는 약물 안전성 정보, 약학정보원(대한약사회 지역 환자안전센터), 03 21 2023
|
|||
| 대한약사저널 |
한약제제
관절질환 <3> 관절 통증 감소 도움되는 계지가출부탕 - 마황 들어간 '의이인탕' 통증에 활용, 최해륭 약사, 2023-02-20
한약제제
관절질환 <2> 풍·습 원인, 면역반응과 염증 유발 - 대방풍탕·방기황기탕 등 활용, 최해륭 약사, 2023-02-13
OTC vs OTC
류마티스관절염 - (건강기능식품) 리프리놀포르테 vs 관절진보원, 고영일 약사, 2023-02-06
한약제제
관절질환 <1> 체액 정체·염증성 물질 발현이 원인 - 미역줄나무 '뇌공등' 관절염 등에 대한 연구중, 최해륭 약사, 2023-02-06
이슈트랜드
염증성장질환(Inflammatory bowel disease, IBD) <2>- 궤양성대장염·크론병 진단 치료법 달라, 순천대 약학대학 최경희 교수, 2022-02-28
이슈트랜드
염증성장질환(Inflammatory bowel disease, IBD) <1>- 영양실조 나타날 수 있으니 주의하세요, 순천대 약학대학 최경희 교수, 2022-02-21
이슈트랜드
염증장질환과 TNF-α 억제제, 숙명여대 약학대학 김현아 교수, 2021-07-26
이슈트랜드
크론병의 약물요법 - 증상경감과 합병증 예방 위해 빠른 염증 억제 필요, 숙명여대 약학대학 김현아 교수, 2021-07-19
이슈트랜드
궤양대장염의 치료 - 염증 억제·질병 완화 유도하는 염증질환 약물요법, 숙명여대 약학대학 김현아 교수, 2021-07-12
이슈트랜드
염증장질환의 병인 - 유전·면역·심리, 이유는 다양하다, 숙명여대 약학대학 김현아 교수, 2021-07-05
이슈트랜드
류마티스관절염 약료의 최신 지견 <2>, 약학정보원 학술정보센터, 2021-01-25
한약제제
계지가출부탕 투여 시 관절 통증 감소 - 보중익기탕, 자음강화탕 등 활용, 최해륭 약사, 2021-01-25
한약제제
류마티스관절염, '풍습'과 관련 깊어 - 대방풍탕, 방기황기탕, 의이인탕 등 활용, 최해륭 약사, 2021-01-18
이슈트랜드
류마티스 관절염 약료의 최신 지견 <1>, 약학정보원 학술정보센터, 2021-01-18
이슈트랜드
류마티스 관절염, 유전적·환경적 요인 발생, 2021-01-11
한약제제
산 중턱에 피어난 류마티스 치료제 <2> - 미역줄나무 Triptolide·celastrol 대표적인 활성성분; 항염증 작용으로 활막 내 대식세포 침투 억제, 2021-01-11
한약제제
산 중턱에 피어난 류마티스 치료제 <1> - 면역세포 유도하는 '미역줄나무', 2021-01-04
이슈트랜드
류마티스 관절염 병태생리의 최신 지견, 2021-01-04
이슈트랜드
건선이 유발하는 '관절염', 치료 방법은?, 2020-11-23
한약제제
건선에 활용되는 한방제는? - 식독에는 '방풍통성산', 어혈형 건선에는 '계지복령환', 최해륭 약사, 2020-11-23
한약제제
전통의학에서의 건선의 증상 및 병리 <2> - 황련해독탕에 합방하는 방제; 청열제·보혈제·구어혈제 병용 시 효과 우수, 최해륭 약사, 2020-11-16
이슈트랜드
건선 치료의 최신 지견 <2>, 약학정보원 학술정보센터, 2020-11-16
이슈트랜드
건선 치료의 최신 지견 <1>, 약학정보원 학술정보센터, 2020-11-09
한약제제
전통의학에서의 건선의 증상 및 병리 <1> - 열 식히는 청열제 사용…황련해독탕 대표적, 최해륭 약사, 2020-11-09
한약제제
건선, 겉이 아닌 근본을 치료해야 - 염증 개선·영양공급 개선 ‘활혈화어제’도 도움, 최해륭 약사, 2020-11-02
이슈트랜드
건선 병태생리의 최신 지견, 약학정보원 학술정보센터, 2020-11-02
|
|||
| 제품설명서 | 보 기 ( 2017-10-10 게시 ) | |||
| 의약품안전성 정보(DUR) |
||||
| 포장단위 (식약처 기준) |
||||
| 저장방법 | ||||
사용자GNB바
컨텐츠
의약품 상세정보

허가정보 ∙ 복약정보
효능 · 효과
성인(18세 이상)
메토트렉세이트를 포함한 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대한 반응이 적절하지 않은 성인의 중등도에서 중증의 활동성 류마티스 관절염의 치료
이전에 메토트렉세이트로 치료받지 않은 성인의 중증의 활동성 및 진행성 류마티스 관절염의 치료
이 약은 단독투여 또는 메토트렉세이트나 다른 DMARDs와 병용투여할 수 있다. 메토트렉세이트에 내약성이 없거나 지속적인 병용투여가 부적절한 경우에는 단독투여한다.
이 약과 메토트렉세이트와의 병용투여는 관절손상 진행속도를 감소시키고(X ‑선측정 결과) 신체활동기능을 향상시킨다.
2. 건선성 관절염
이전에 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대한 반응이 적절하지 않은 성인의 활동성 및 진행성 건선성 관절염의 치료. 이 약은 다발성, 대칭성 아형을 지닌 환자의 말초 관절손상 진행속도를 감소시키고(X ‑선측정 결과), 신체활동기능을 향상시킨다.
3. 축성 척추관절염
(1) 강직성 척추염
기존 치료에 대한 반응이 적절하지 않은 성인의 중증의 강직성 척추염의 치료
(2) 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염
방사선학적 검사에서 강직성 척추염이 확인되지 않으나, 상승된 CRP 수치 및/또는 MRI상 객관적인 염증의 징후를 보이는 중증 축성 척추관절염의 치료. 이 약은 비스테로이드성항염증제(NSAIDs) 약물에 대한 반응이 적절하지 않거나, 불내성인 환자에 사용한다.
4. 크론병
코르티코스테로이드제나 면역억제제 등의 치료에 반응을 나타내지 않거나, 내약성이 없는 경우 또는 이러한 치료방법이 금기인 중등도에서 중증의 활성 크론병의 치료
유도 요법의 경우 이 약은 코르티코스테로이드제와 병용투여한다. 코르티코스테로이드제에 내약성이 없거나, 지속적인 병용투여가 부적절한 경우 단독투여할 수 있다.
5. 건선
전신치료 또는 광선요법이 필요한 성인의 중등도에서 중증의 만성 판상 건선의 치료
6. 궤양성 대장염
코르티코스테로이드 및 6 ‑MP(6 ‑mercaptopurine) 또는 AZA(azathioprine)를 포함한 통상적인 치료에 대해 반응이 적절하지 않거나 내약성이 없는 경우 또는 이러한 치료방법이 금기인 성인의 중등도에서 중증의 활성 궤양성 대장염의 치료
7. 베체트 장염
스테로이드 또는 면역억제제 등의 통상적인 치료에도 적절한 반응이 나타나지 않는 베체트 장염의 치료
8. 화농성 한선염
기존의 전신 요법에 적절한 반응을 나타내지 않는 중등도에서 중증의 활성 화농성 한선염의 치료
9. 포도막염
코르티코스테로이드에 적절한 반응을 나타내지 않은 성인의 비 ‑감염성 중간 포도막염, 후포도막염 및 전체포도막염의 치료
소아
1. 소아 크론병(6 ‑ 17세)
일차 영양요법, 코르티코스테로이드, 면역조절제 등의 치료에 적절한 반응을 나타내지 않거나, 내약성이 없는 경우 또는 이러한 치료방법이 금기인 소아 환자(6 ‑ 17세)에서 중등도에서 중증의 활성 크론병의 치료
2. 소아 특발성 관절염
(1) 다관절형 소아 특발성 관절염
하나 이상의 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대해 부적절한 반응을 보인 2세 이상의 소아 및 청소년에서 활성 다관절형 소아 특발성 관절염의 치료
(2) 골부착부위염 관련 관절염
기존 치료에 적절한 반응을 나타내지 않거나 내약성이 없는 6세 이상 어린이 및 청소년의 활성 골부착부위염 관련 관절염의 치료
3. 소아 판상 건선
국소 치료 및 광선요법에 적절한 반응을 나타내지 않거나 해당 치료가 부적절한 4세 이상의 어린이 및 청소년의 중증 만성 판상 건선의 치료
용법 · 용량
환자가 자가 주사하는 것이 적절하며 필요시 치료 추적이 가능하다고 의사가 판단하는 경우, 환자는 주사방법에 대한 교육을 받은 후 이 약을 자가 주사할 수 있다.
성인
1. 류마티스 관절염
성인 류마티스 관절염 환자에 대한 이 약의 권장 용량은 아달리무맙 40 mg을 2주에 1회 피하주사한다. 이 약을 투여하는 동안 메토트렉세이트의 병용투여를 유지한다.
글루코코르티코이드, 살리실산염, 비스테로이드성항염증제(NSAIDs), 진통제, 다른 DMARDs의 병용투여를 유지할 수 있다.
단독요법의 경우 이 약에 대한 반응이 감소한 환자는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하면 유용한 효과를 얻을 수 있다.
2. 건선성 관절염, 강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염
건선성 관절염, 강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염 환자에 대한 이 약의 권장 용량은 아달리무맙 40 mg을 2주에 1회 피하주사한다.
상기 효능 ▪효과에 대해 임상적인 반응은 보통 치료 12주 이내에 나타나며, 이 기간 내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
3. 성인 크론병
성인 중등도에서 중증 크론병에 대한 이 약의 권장 용량은 첫 주에 아달리무맙 80 mg을 투여하고 첫 투여 후 2주 후에 40 mg을 투여한다. 빠른 효과를 얻어야 할 필요가 있는 경우에는, 유도 요법동안 이상 반응에 대한 위험성이 증가한다는 것을 알리고, 첫 주에 160 mg을 투여하고 첫 투여 후 2주 후에 80 mg을 투여할 수 있다. 유도 요법의 경우 이 약은 코르티코스테로이드제와 병용투여한다. 코르티코스테로이드제에 내약성이 없거나, 지속적인 병용투여가 부적절한 경우 단독투여할 수 있다.
유도 요법 후, 아달리무맙 40 mg을 2주에 1회 피하주사한다. 만약 투여를 중지하고 재발의 증상과 징후가 나타나면 이 약을 재투여 할 수 있다. 8주 이상의 투여 중지 후 재투여에 대한 경험은 거의 없다.
유지 요법 동안 임상 지침에 따라 코르티코스테로이드제의 투여를 서서히 줄일 수 있다.
이 약에 대한 반응이 감소한 환자는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하면 유용한 효과를 얻을 수 있다.
4주까지 반응을 보이지 않는 환자의 경우 12주까지 투여하여 반응을 나타낼 수 있다. 이 기간 내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
4. 건선
성인에 대한 이 약의 권장 용량은 첫 회에 아달리무맙 80 mg을 피하로 투여하고, 이어서 첫 투여 후 1주일 후에 40 mg을 격주로 투여한다.
16주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
충분한 반응이 나타나지 않는 환자는 40mg 매주 투여 또는 80 mg 격주 투여로 증량하여 유용한 효과를 얻을 수 있다. 증량 후 충분한 반응이 나타나지 않는 환자의 경우에는 이 약의 지속적 40 mg 매주 투여 또는 80 mg 격주 투여의 유용성과 위험성을 신중히 재고한다.
5. 궤양성 대장염
성인 중등도에서 중증의 궤양성 대장염에 대한 이 약의 권장 용량은 아달리무맙 160 mg을 투여하고 첫 투여 2주 후에 80 mg을 투여하는 것이다. 유도 요법 후 40 mg을 격주로 피하주사한다.
유지 요법 동안 임상 지침에 따라 코르티코스테로이드제의 투여를 서서히 줄일 수 있다.
이 약에 대한 반응이 감소한 환자 중 일부는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하여 유용한 효과를 얻을 수 있다.
지금까지 나온 데이터에 따르면 임상 반응은 대체로 투여 2 ‑8주 이내에 도달한다. 이 기간 내에 반응을 보이지 않은 환자의 경우 투여 지속여부를 신중히 재고한다.
6. 베체트 장염
성인에 대한 이 약의 권장 용량은 첫 회에 160 mg을 피하로 투여하고 이어서 첫 투여 2주 후에 80 mg을 투여한다. 첫 투여 4주 후부터는 40 mg을 격주로 투여한다.
12주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
7. 화농성 한선염
이 약의 권장용량은 첫 회에 160 mg을 피하로 투여하고, 이어서 첫 투여 2주 후에 80 mg을 투여한다. 첫 투여 4 주 후 부터는 40 mg을 매주 투여 또는 80 mg을 격주 투여한다. 필요 시, 이 약의 치료 기간 동안 항생제를 계속 투여할 수 있다.
12주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
치료를 중단해야 하는 경우, 40mg 매주 투여 또는 80 mg 격주 투여로 재투여할 수 있다.
8. 포도막염
성인에 대한 이 약의 권장 용량은 첫 회 80 mg 투여하고 이어서 첫 투여 1주 후부터 40mg을 격주로 투여한다.
이 약은 단독 또는 코르티코스테로이드 또는 면역억제제 (생물학적 면역억제제 제외)와 병용하여 사용할 수 있다. 코르티코스테로이드는 임상 경험에 따라 양을 줄일 수 있다.
소아
1. 소아 크론병
6세 이상의 소아 크론병 환자에서 이 약의 권장 투여 용량은 체중을 기반으로 한다. 이 약은 피하 투여한다.
|
체중 (kg) |
유도 용량 |
유지 용량 |
|
40 kg 미만 |
ㆍ 첫 주에 40mg 투여하고 첫 투여 후 2주 후에 20mg 투여 ㆍ 빠른 효과를 얻어야 할 필요가 있는 경우에는, 고용량의 유도요법 동안 이상사례에 대한 위험성이 증가한다는 것을 알리고 아래 용량을 투여할 수 있다. - 첫 주에 80mg 투여하고, 첫 투여 후 2주 후에 40mg 투여 |
20mg 격주 투여 |
|
40 kg 이상 |
ㆍ 첫 주에 80mg 투여하고 첫 투여 후 2주 후에 40mg 투여 ㆍ 빠른 효과를 얻어야 할 필요가 있는 경우에는, 고용량의 유도요법 동안 이상사례에 대한 위험성이 증가한다는 것을 알리고 아래 용량을 투여할 수 있다. - 첫 주에 160mg 투여하고, 첫 투여 후 2주 후에 80mg 투여 |
40mg 격주 투여 |
◦ 40 kg 미만: 20 mg 매주 투여
◦ 40 kg 이상: 40 mg 매주 투여 또는 80 mg 격주 투여
12주까지 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
2. 소아 특발성 관절염
(1) 다관절형 소아 특발성 관절염
이 약은 2세 미만 소아 또는 체중 10 kg 미만 환자를 대상으로 연구된 바 없다.
이 약의 권장 투여 용량은 체중을 기반으로 한다. 이 약은 메토트렉세이트와 병용하여 투여한다. 메토트렉세이트에 내약성이 없거나 메토트렉세이트와 함께 지속적으로 투여하는 것이 부적절할 경우에는 이 약을 단독요법으로 투여할 수 있다.
|
체중 (kg) |
용량 |
|
10 kg 이상 30 kg 미만 |
20mg 격주 투여 |
|
30 kg 이상 |
40mg 격주 투여 |
(2) 골부착부위염 관련 관절염
6세 이상의 골부착부위염 관련 관절염 환자에 대한 이 약의 권장 투여 용량은 체중을 기반으로 한다.
|
체중 (kg) |
용량 |
|
15 kg 이상 30 kg 미만 |
20mg 격주 투여 |
|
30 kg 이상 |
40mg 격주 투여 |
3. 소아 판상 건선
소아 판상 건선에 대한 이 약의 권장 투여 용량은 체중을 기반으로 한다. 16주 이후에도 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
|
체중 (kg) |
용량 |
|
15 kg 이상 30 kg 미만 |
20mg을 처음 2회는 매주 피하주사하고, 이후에는 격주 투여 |
|
30 kg 이상 |
40mg을 처음 2회는 매주 피하주사하고, 이후에는 격주 투여 |
소아 판상 건선으로 4세 미만의 어린이에게 이 약을 사용한 적은 없다.
사용상의 주의사항
1. 경고
세균성, 진균성, 침습적 진균(파종성 또는 폐외 히스토플라스마증, 아스페르길루스증, 콕시디오이데스진균증), 바이러스성으로 인한 중대한 감염, 기생충에 의한 감염 또는 기타 기회감염이 TNF 저해제를 투여 받고 있는 환자들에게서 보고되었다. 또한 패혈증, 드물게 결핵, 칸디다증, 리스테리아증, 레지오넬라증 및 뉴모시스티스(pneumocystis)가 이 약을 포함한 TNF 저해제의 사용에서 보고되었다. 감염과 관련된 입원 또는 치명적인 결과가 보고되었으며, 심각한 감염은 대부분 선행 질환과 함께 감염을 일으키기 쉬운 면역억제제를 병용 투여한 환자에서 발생했다.
⓵ 심각한 감염
이 약을 투여받고 있는 환자에게 심각한 감염의 위험이 증가되는 경우가 임상시험에서 나타났으며, 시판 후 조사에서 이러한 사실을 뒷받침하는 보고가 있었다. 특히 폐렴, 신우신염, 패혈성 관절염, 패혈증과 같은 심각한 감염이 보고되었다.
⓶ 결핵
이 약을 투여받고 있는 환자에게서 결핵(재활성화 및 발병 포함)이 보고되었으며, 폐결핵 및 폐외결핵(예, 파종성)이 포함되었다.
이 약으로 치료를 시작하기 전 모든 환자의 활동성 또는 비활동성(잠복성) 결핵 감염에 대해 평가해야 한다. 이 평가에는 개인의 결핵 병력 또는 과거에 활동성 결핵 환자에 노출되었을 가능성과 과거 및 현재 면역억제요법에 대한 자세한 의학적 평가가 포함되어야 한다. 모든 환자에서 투베르쿨린 피부 검사와 흉부 X ‑선과 같은 적절한 스크리닝 시험을 실시해야 한다(국가별 권장사항을 적용할 수 있음).
특히 결핵이 널리 유행되는 지역에서 이민오거나 혹은 그곳을 여행한 환자들 또는 활동성 결핵 환자와 긴밀한 접촉이 있었던 환자들에서 진단되지 않은 잠복성 결핵이 가능하다는 것을 고려해야만 한다.
활동성 결핵으로 진단된 경우에는 이 약의 투여를 시작해서는 안 된다.
잠복성 결핵으로 진단된 경우에는, 이 약의 투여 시작 전 항결핵 예방요법과 함께 적절한 치료를 시작해야 하며, 국가별 권장사항에 따라야 한다. 결핵 테스트 결과가 음성일지라도 결핵의 중대한 위험인자를 갖고 있거나 여러 위험인자가 있는 환자 및 과거 활성 또는 잠복성 결핵의 병력이 있으나 적절한 치료가 확인되지 않은 환자 또한 이 약의 투여 시작 전에 항결핵 예방요법을 고려해야한다. 이 환자에 대한 항결핵 치료 시작은 잠복성 결핵 감염에 대한 위험과 항결핵 치료의 위험 두 가지를 모두 고려한 후에 결정되어야만 한다. 만약 필요하다면, 결핵치료의 경험이 있는 의사와 상담하여야만 한다.
잠복성 결핵 감염 환자의 항결핵 치료는 이 약을 투여받는 환자들의 결핵 재활성화 위험을 감소시킨다. 항결핵 예방요법에도 불구하고, 이 약을 투여 받는 동안 활동성 결핵이 발생한 경우가 있었다. 또한 잠복성 결핵 결과가 음성이었던 환자에서 이 약을 투여받고 활성 결핵이 발생하고, 활성 결핵 치료가 성공적으로 실시된 몇몇 환자에서도 TNF 저해제로 투여받는 동안 활성 결핵이 다시 나타난 바가 있다.
이 약으로 치료받는 환자는 잠복성 결핵이 위음성일 수 있으므로, 활성 결핵의 증상 및 징후가 모니터링되어야 한다. 투베르쿨린 위음성(false negative) 결과의 위험은 중증 질환자나 면역기능저하환자에서 특히 고려해야한다.
만약 이 약으로 치료하는 동안이나 치료 후 결핵을 암시하는 증상/징후(예. 지속성 기침, 쇠약/체중감소, 미열, 무기력)가 나타나는 경우 의사에게 즉시 알리도록 환자를 교육해야 한다.
⓷ 기타 기회감염
침습적 진균 감염을 포함한 기회감염이 이 약을 투여 받고 있는 환자들에서 관찰되었다. 이 감염들은 TNF 저해제를 투여 받고 있는 환자들에게서 일관되게 나타나지는 않았으며, 이는 적절한 치료의 지연을 야기하였고, 때로는 치명적인 결과를 초래하였다.
TNF 저해제를 투여받고 있는 환자들은 히스토플라스마증, 콕시디오이데스진균증, 또는 분아균증(blastomycosis), 아스페르길루스증, 칸디다증 및 기타 기회감염들에 좀 더 감수성이 높다. 발열, 권태감, 체중 감소, 발한, 기침, 호흡곤란 및/또는 폐 침윤, 또는 기타 심각한 (쇼크를 동반하거나 동반하지 않은) 전신질환이 나타난 환자는 즉시 진단 평가를 위한 의학적 조치를 취해야 한다.
진균증이 풍토병인 지역에 머물거나 여행한 환자의 경우, 전신적인 진균 감염의 징후가 나타나면, 침습적인 진균 감염을 의심해야 한다. 환자들은 히스토플라스마증 및 기타 침습적 진균 감염의 위험이 있으므로, 임상 의사들은 병원균이 확인되기 전까지 실험적 항진균 치료를 고려해야 한다. 활성 감염 상태인 몇몇의 환자들은 히스토플라스마증의 항원 및 항체 검사에서 음성을 나타낼 수 있다. 이러한 환자들에게 실험적 항진균 치료제의 투여는, 침습적 진균 감염의 치료 및 진단에 전문가인 의사와 상담하여 결정되어야 하며, 심각한 진균 감염의 위험 및 항진균 치료의 위험을 모두 고려하여야 한다. 심각한 진균 감염이 발생한 환자 또한 감염이 조절되기 전까지 TNF 저해제의 사용 중단이 고려되어야 한다.2) 악성종양과 림프증식성 질환
TNF 저해제에 대한 임상시험의 대조 시험동안 TNF 저해제를 투여받은 환자에서 대조군 환자에 비해 더 많은 림프종 사례가 관찰되었다. 그러나 발생이 드물었고 위약군 환자의 추적조사기간이 TNF 저해제를 투여받은 환자에 비해 더 짧았다. 더욱이 오래 지속된, 매우 활동성인, 염증성 질환을 가진 류마티스 관절염 환자는 림프종의 기저 위험(background risk)이 증가되어 있고 이는 위험 추정을 어렵게 한다. 이 약을 이용한 장기간 공개 임상시험에서 악성종양의 전체 발현율은 나이, 성별, 인종이 맞춰진 전체 인구에서 예상되는 발현율과 유사했다. 현재의 지식으로 TNF 저해제를 투여받는 환자에서 림프종 또는 다른 악성종양의 발생 위험 가능성을 배제할 수 없다.
TNF 저해제를 투여받는 어린이와 청소년들에서 악성종양이 보고되었고, 일부는 치명적이었다. 이 보고된 경우 중 대략 절반의 경우는 호지킨 및 비호지킨 림프종을 포함한 림프종이었다. 나머지 다른 경우들에서는 여러 다양한 악성 종양들이 나타났으며, 일반적으로 면역억제와 연관되어 드물게 나타나는 악성종양도 포함되었다. 악성종양은 중간값으로 치료 30개월 후에 나타났으며, 대부분의 환자가 면역억제제를 병용 투여하고 있었다. 이는 시판 후 조사에서 보고되었으며, 등록과 자발적인 시판 후 보고서를 포함한 여러 경로에서 나타났다.
이 약을 투여받은 환자에서, Hepatosplenic T ‑cell lymphoma(HSTCL)가 시판후 조사에서 매우 드물게 보고되었고, 공격적 림프종(종종 치명적임)이 드물게 확인되었다. 대부분의 환자들은 염증성대장질환의 치료를 위하여 아자티오프린 또는 6 ‑메르캅토푸린의 병용 투여는 물론이고 이전에 인플리시맵으로 치료를 받았었다. 이 약과 아자티오프린 또는 6 ‑메르캅토푸린의 병용 투여 시의 잠재적인 위험성을 주의깊게 고려해야 한다. 이 약 복용환자에서 HSTCL의 발생 위험 가능성은 배제할 수 없다
악성종양의 병력이 있는 환자를 포함하거나 이 약 투여 중 악성종양이 발생한 환자에게 투여를 지속한 시험은 실시되지 않았다. 따라서 이러한 환자군에 투여를 고려시 추가적인 주의를 기울여야 한다. 모든 환자, 특히 과다한 면역억제제 치료의 병력이 있는 환자나, PUVA 치료의 병력이 있는 건선 환자들은 이 약의 투여 시작 전 및 투여 기간 동안 비흑색종 피부암 존재 여부에 대한 검사를 받아야 한다.
TNF 저해제를 류마티스 관절염과 다른 적응증에서 사용하여 급성 및 만성 백혈병이 나타난 경우가 보고되었다. 류마티스 관절염이 있는 환자들은 TNF 저해제를 투여받지 않더라도 다른 사람들에 비해 백혈병이 발병될 확률이 (2배까지) 높을 수 있다.
중등증에서 중증의 만성 폐쇄성 폐질환(COPD) 환자들을 대상으로 다른 TNF 저해제인 인플릭시맵을 사용하여 실시한 탐색적 임상시험에서, 현재 흡연중이거나 기존에 흡연하였던 사람의 경우 비교군의 환자보다 인플릭시맵 치료를 받은 환자군에서 악성 종양이 많이 보고되었다. 과도한 흡연으로 인해 악성종양의 위험성이 증가한 환자는 물론 만성 폐쇄성 폐질환(COPD) 환자에 대한 TNF ‑α 저해제 치료 고려 시 주의가 필요하다.
이 약이 형성이상증이나 대장암의 발생 위험에 영향을 미치는지 여부는 현재 알려지지 않았다. 형성이상증 또는 대장암종의 위험이 증가되었거나(예, 오래 지속되는 궤양성 대장염 또는 원발성 경화성 담관염), 형성이상증 또는 대장암종의 병력이 있는 모든 궤양성 대장염 환자들은 치료 시작 전 및 치료 중에 주기적으로 형성이상증에 대해 확인해야한다. 대장 내시경검사 및 생검 등 국가별 권장사항에 따라 평가한다.
3) B형 간염 재활성화
이 약을 포함한 TNF 저해제의 사용으로 B형 간염 만성 보균자에서 병증이 재활성화 될 수 있으며, 일부는 치명적이었다. 이 보고내용의 대부분은 다른 면역억제제를 함께 투여받는 환자에게서 나타났으며, 이는 또한 B형 간염 재활성화를 일으킬 수 있다. B형 간염의 감염 위험이 있는 환자는 이 약을 투여 받기 전 감염 여부에 대한 검사를 하여야 한다. B형 간염 보균자로 확인된 환자에게 TNF 저해제를 처방하는 경우, 의사는 주의를 기울여야 한다. B형 간염 보균자이고, TNF 저해제의 치료를 요하는 환자는 이 약을 투여 받는 동안과 투여가 끝난 후 수 개월 동안 감염의 증상/징후를 철저히 관찰해야 한다. B형 간염 보균자의 재활성화를 방지하기 위해 항바이러스제와 TNF 저해제를 함께 사용하는 것에 대한 적절한 치료법은 알려진 바 없다. B형 간염이 재활성화 된 경우 이 약의 투여를 중지하고 적절한 치료와 함께 항바이러스제 치료를 시작해야 한다.
4) 신경학적 반응
이 약을 포함한 다른 TNF 저해제는 드물게, 다발성 경화증 및 시신경염을 포함한 중추신경계 탈수초성질환(demyelinating disease)과 길랑 바레 증후군(Guillian Barre syndrome)을 포함한 말초 탈수초성질환의 임상 증상 및/또는 방사선 증거의 새로운 발현 혹은 악화와 관련이 있다. 중추신경계 및 말초신경계 탈수초성질환의 기왕력이 있거나 최근에 발현된 환자에게 이 약의 투여를 고려하는 경우 주의를 기울여야 한다. 이러한 이상이 진행되면 이 약의 중단을 고려하여야 한다.
중등도 포도막염과 중추 신경계 탈수초성장애 간의 연관성은 알려져있다. 비 ‑감염성 중등도 포도막염 환자에서 이 약 치료 전 중추신경계 탈수초성 장애 여부를 측정하는 신경학적 평가를 하여야 한다.
2. 다음 환자에는 투여하지 말 것
1) 이 약 또는 이 약의 성분에 과민증인 환자
2) 활동성 결핵 또는 패혈증, 기회감염과 같은 다른 중증 감염이 있는 환자(경고항 참조)
3) 중등도 내지 중증의 심부전(NYHA class III/IV) 환자
3. 이상반응
1) 이 약은 치료적 확증 대조 및 공개 임상시험을 통해 60개월 이상에 걸쳐 9,506명의 환자에 대하여 연구되었다. 이 임상시험들에는 단기 및 장기 질병상태를 보이는 류마티스 관절염 환자뿐만 아니라 건선성 관절염 및 축성 척추관절염(강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염) 환자 및 크론병 환자, 궤양성 대장염 환자 및 건선 환자, 화농성 한선염 환자, 포도막염 환자와 소아 특발성 관절염 (다관절 소아 특발성 관절염 및 골부착부위염 관련 관절염) 환자가 포함되었다. 표 1의 자료는 대조 임상시험 기간 동안 이 약을 투여받은 6,089명의 환자와 위약 또는 활성 대조약을 투여받은 3,801명의 환자를 포함한 주요 대조 임상시험 결과를 근거로 한 것이다. 주요 임상시험 중 이중 눈가림, 대조 임상시험 기간 동안 이상사례로 인해 투여를 중단한 환자의 비율은 이 약 투여군이 5.9%이고 대조군이 5.4%였다.
일반적으로 소아환자에서의 이상사례는 성인 환자에서 나타난 이상사례의 빈도 및 타입과 유사하였다.
이 약을 이용한 대조 임상시험에서 가장 빈번히 나타났던 이상사례들 중 하나를 근거로 약 13%의 환자들이 주사부위반응을 경험할 것으로 예상할 수 있다.
아래 표 1은 주요 임상시험에서 임상적 및 실험실 검사 모두 적어도 이 약과 인과관계의 가능성이 있는 약물이상반응을 발현부위와 빈도별(매우 자주 ‑1/10 이상 ; 자주 ‑1/100 이상 및 1/10 미만 ; 때때로 ‑1/1,000 이상 및 1/100 미만 ; 드물게 ‑1/1,000 미만)로 요약하였다. 각각의 발현빈도 그룹 안에, 약물이상반응은 심각성이 감소하는 순서로 배열되었다. 이 약의 여러 적응증에서 가장 자주 나타난 약물이상반응들이 포함되었다. 신체기관 열에서 (*)로 표시된 부분은 1. 경고, 4. 일반적 주의 항에서 자세한 내용을 확인할 수 있다.
2) 베체트 장염 : 일본의 베체트 장염 연구에서의 안전성 프로파일은 이미 연구된 이 약의 안전성 프로파일과 유사하였다.
3) 화농성 한선염: 이 약으로 매주 치료받은 화농성 한선염 환자의 안전성 프로파일은 이미 알려진 이 약의 안전성 프로파일과 유사하였다.
4) 포도막염: 이 약으로 치료받은 비 ‑감염성 포도막염 환자의 안정성 프로파일은 이미 알려진 이 약의 안전성 프로파일과 유사하였다.
<표 1. 임상시험 중 약물이상반응> <! ‑ ‑표 ‑ ‑><! ‑ ‑표 ‑ ‑><! ‑ ‑표 ‑ ‑>
|
신체기관 |
빈도 |
약물이상반응 |
|
감염 |
매우 자주 |
기도 감염(상기도 및 하기도 감염, 폐렴, 부비동염, 인두염, 비인두염, 폐렴 헤르페스 바이러스 포함) |
|
자주 |
전신 감염(패혈증, 칸디다증, 인플루엔자 포함), 장내 감염(위장염바이러스 포함), 피부 및 연조직 감염(손발톱주위염, 연조직염, 괴사성 근막염, 바이러스성 수막염, 게실염, 상처 감염, 농가진, 괴사근막염, 대상포진), 귀 감염, 구내감염(단순포진, 구강 헤르페스, 치아 감염 포함), 생식기 감염(진균성 외음부 및 질 감염 포함), 비뇨기계 감염(신우신염 포함), 진균 감염, 관절 감염 | |
|
때때로 |
기회감염과 결핵(콕시디오이데스진균증, 히스토플라스마증, 계형결핵균 복합체 감염), 신경 감염(바이러스성 수막염 포함), 눈 감염, 박테리아 감염 | |
|
양성, 악성 및 미확인 신생물(낭종 및 폴립 포함)(*) |
자주 |
양성종양, 악성흑색종을 제외한 피부암(기저세포암, 편평세포암을 포함) |
|
때때로 |
림프종, 실질기관종양(유방암, 폐종양, 갑상선 종양 포함), 악성흑색종 | |
|
혈액계 및 림프계 이상(*) |
매우 자주 |
백혈구감소증(호중구감소증, 무과립구증 포함), 빈혈 |
|
자주 |
저혈소판증, 백혈구증가증 | |
|
때때로 |
특발성 혈소판감소성 자반증 | |
|
드물게 |
범혈구감소증 | |
|
면역계 이상 |
때때로 |
과민반응, 알레르기(계절성 알레르기 포함) |
|
대사 및 영양계 이상 |
매우 자주 |
지질증가 |
|
자주 |
저칼륨혈증, 고뇨산혈증, 혈중나트륨농도이상, 저칼슘혈증, 고혈당증, 저인산혈증, 탈수증 | |
|
정신계 이상 |
때때로 |
감정이상(우울증 포함), 불안, 불면증 |
|
신경계 이상 |
매우 자주 |
두통 |
|
자주 |
피부지각이상(감각저하증 포함), 편두통, 신경뿌리압박 | |
|
때때로 |
떨림, 신경병증 | |
|
드물게 |
다발성경화증 | |
|
눈의 이상 |
자주 |
시각장애, 결막염, 눈꺼풀염, 눈부종 |
|
때때로 |
복시 | |
|
귀 및 미로 이상 |
자주 |
어지럼증 |
|
때때로 |
난청, 이명 | |
|
심장 이상 |
자주 |
빈맥 |
|
때때로 |
부정맥, 울혈성 심부전 | |
|
드물게 |
심정지 | |
|
혈관 이상 |
자주 |
고혈압, 홍조, 혈종 |
|
때때로 |
혈관동맥 폐색증, 혈전정맥염, 대동맥류 | |
|
호흡, 흉부 및 종격 이상 |
자주 |
기침, 천식, 호흡곤란 |
|
때때로 |
만성 폐쇄성 폐질환, 간질성 폐질환, 폐렴 | |
|
위장관계 이상 |
매우 자주 |
복통, 구역 및 구토 |
|
자주 |
위장관출혈, 소화불량, 위식도역류 질환, 건조증후군 | |
|
때때로 |
췌장염, 연하곤란, 얼굴부종 | |
|
간 및 담즙계 |
매우 자주 |
간 효소 상승 |
|
때때로 |
담낭염, 담석증, 빌리루빈 상승, 지방간 | |
|
피부 및 피하조직 이상 |
매우 자주 |
발진(박리성 발진 포함) |
|
자주 |
가려움증, 두드러기, 타박상(자색반증 포함), 피부염(습진 포함), 조갑박리증, 다한증 | |
|
때때로 |
야간발한, 흉터 | |
|
근골격계 및 결합조직 이상 |
매우 자주 |
근골격통 |
|
자주 |
근경련(혈중크레아틴인산활성효소 증가 포함) | |
|
때때로 |
횡문근융해, 전신홍반루푸스 | |
|
신장 및 비뇨 이상 |
자주 |
혈뇨, 신기능 손상 |
|
때때로 |
야뇨증 | |
|
생식기계 및 유방 이상 |
때때로 |
발기부전 |
|
전신이상 및 투여 부위 이상 |
매우 자주 |
주사부위반응(주사부위 홍반 포함) |
|
자주 |
흉통, 부종 | |
|
때때로 |
염증 | |
|
임상 실험실 검사 |
자주 |
응고 및 출혈이상(활성화부분트롬보플라스틴시간 지연 포함), 자가항체시험 양성(이중나선DNA항체 포함), 혈중 락트산탈수효소 증가 |
|
상해 및 중독 |
자주 |
치유 부전 |
이 약으로 치료를 받은 류마티스 관절염 환자에게 5% 미만의 드문 발생률로 나타난 기타 중대한 이상사례는 다음과 같다.
전신 : 발열, 감염, 극심한 통증, 골반 통증, 패혈증, 수술, 가슴통증, 결핵의 재발
심혈관계 : 부정맥, 심방 세동, 심혈관 장애, 흉통, 울혈성 심부전, 관상동맥 이상, 심정지, 고혈압성 뇌병증, 심근경색증, 심계항진, 심낭삼출, 심낭염, 실신, 빈맥, 혈관 장애
콜라겐 질환 : 홍반성 루푸스 증후군
소화기계 : 담낭염, 담석증, 식도염, 위염, 위장장애, 위장출혈, 간 괴사, 구토
내분비계 : 부갑상선 장애
혈액계 : 무과립구증, 과립구감소증, 백혈구감소증, 림프종 유사반응, 범혈구감소증, 적혈구증가증
대사 및 영양장애 : 탈수증, 치유 이상, 케톤증, 파라프로테인혈증, 말초부종
근골격계 : 관절염, 뼈장애, 뼈골절(자연적인 경우가 아닌), 골괴사, 관절장애, 근육경련, 중증근무력증, 화농성 관절염, 윤활막염, 힘줄 장애
종양 : 샘종, 유방 ◦ 위장관 ◦ 피부 ◦ 비뇨생식과 같은 암종 및 기타(림프종, 흑색종)
신경계 : 착란, 다발성 경화증, 감각이상, 경막하혈종, 떨림
호흡기계 : 천식, 기관지연축, 호흡곤란, 폐 장애, 폐기능 감소, 흉막 삼출, 폐렴
피부 및 부속물 : 연조직염, 얕은 연조직염, 대상포진
특수감각 : 백내장
혈전증 : 다리혈전
비뇨생식기계 : 방광염, 신장 결석, 월경 장애, 신우신염
5) 주사부위반응 : 성인 및 소아에 대한 주요 대조 임상시험에서 이 약을 투여한 환자의 12.9%가 이 약을 투여했을 때 가장 흔히 나타난 이상사례인 주사부위반응(홍반 및/또는 가려움증, 출혈, 통증 또는 부종)을 나타낸 반면, 위약 또는 활성 대조군을 투여한 환자에서는 7.2%에서 이러한 반응이 나타났다. 주사부위반응은 일반적으로 투여 중단이 필요하지는 않았다.
6) 감염 : 성인 및 소아에 대한 주요 대조 임상시험에서, 감염률은 이 약 투여군이 1.51/환자 ‑년(patient year)이고 위약 및 활성 대조약 투여군은 1.46/환자 ‑년이었다. 감염은 주로 비인두염, 상기도 감염 및 부비동염으로 구성되었다. 대부분의 환자들은 감염이 소실된 후 이 약을 계속 투여하였다. 심각한 감염 발생률은 이 약 투여군이 0.04/환자 ‑년이고 위약 및 활성 대조약 투여군은 0.03/환자 ‑년이었다. 이 약을 이용한 성인 및 소아에 대한 대조 및 공개 임상시험에서 심각한 감염(드물게 발생한 치명적 감염을 포함)이 보고되었으며, 이는 결핵(속립성 및 폐 이외 부위 포함) 및 침습적 기회 감염(예. 파종성 히스토플라스마증, 폐포자충 폐렴, 아스페르길루스증 및 리스테리아증)을 포함한다. 결핵 사례의 대부분은 치료 시작 후 최초 8개월 이내에 발생하였으며, 이는 잠복 질환의 재발을 반영하는 듯하다.
7) 악성종양과 림프증식성 질환 : 소아 특발성 관절염(다관절형 소아 특발성 관절염 및 골부착부위염 관련 관절염) 환자를 대상으로 655.6 환자 ‑년 노출된 249명에서의 임상시험 동안 악성종양이 관찰되지 않았다. 또한, 소아 크론병 환자를 대상으로 498.1환자 ‑년 노출된 192명에서의 임상시험 동안 악성종양이 관찰되지 않았다.
판상 건선 소아 환자를 대상으로 80.0 환자 ‑년 노출된 77명의 소아 환자에서의 임상시험 동안 악성 종양이 관찰되지 않았다. 중등도에서 중증의 류마티스 관절염, 건선성 관절염, 축성 척추관절염(강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염), 크론병, 궤양성대장염, 화농성 한선염, 건선, 포도막염 환자를 대상으로 12주 이상 실시한 성인에 대한 주요 대조 임상시험에서 림프종, 비흑색종 피부암 이외에도 악성종양이 투여군 5,291명 중에서 6.8/1,000 환자 ‑년, 대조군 3,444명 중에서 6.3/1,000 환자 ‑년의 비율로 생성되었다(치료기간의 중간값은 이 약의 투여군에서는 4.0개월, 대조군에서는 3.8개월이었다). 비흑색종 피부암은 투여군에서 8.8/1,000 환자 ‑년 및 대조군에서 3.2/1,000 환자 ‑년의 비율로 생성되었다. 피부암중에서 편평세포암종은 투여군에서 2.7/1,000 환자 ‑년, 대조군에서 0.6/1,000 환자 ‑년의 비율로 생성되었다. 림프종의 비율은 투여군에서 0.7/1,000 환자 ‑년, 대조군에서 0.6/1,000 환자 ‑년이었다.
대조 임상시험과 진행 중 또는 완료된 공개 연장 시험에서, 림프종 및 비흑색종 피부암 이외에 관찰된 악성종양의 발생률은 약 8.5/1,000 환자 ‑년이다. 비흑색종 피부암의 관찰된 비율은 약 9.6/1,000 환자 ‑년이며, 림프종이 관찰된 비율은 약 1.3/1,000 환자 ‑년이다. 이 임상시험기간의 중앙값은 약 3.3년이며, 최소 1년 동안 이 약을 투여받은 6,427명의 환자들 또는 26,439.6 환자 ‑년 투여를 초과하는 치료 시작 1년 이내에 악성종양으로 발전한 환자들을 포함했다.
2003년 1월부터 2010년 12월까지의 시판 후 경험 중 주로 류마티스 관절염 환자에서 보고된 악성종양의 발생 비율은 약 2.7/1,000 환자 ‑년 이었다. 비흑색종 피부암과 림프종의 발생 비율은 각각 약 0.2 및 0.3/1,000 환자 ‑년이었다.
8) 자가항체 : 류마티스 관절염에 대한 5건의 임상시험 중 여러 시점에서 혈청 내 자가항체에 대한 검사를 실시했다. 이러한 적절히 디자인된 대조 임상시험에서 기저 항 핵항체(ANA, anti ‑nuclear antibody)가 음성이었던 환자 중 이 약 투여군의 11.9%와 위약 및 활성 대조군의 8.1%에서 24주째 양성을 나타냈다. 모든 류마티스 관절염과 건선성 관절염에 대해 이 약을 투여받은 3,441명 중 2명에서 새롭게 발생된 루푸스양 증후군임을 시사하는 임상 증상이 나타났으며 이 환자는 투여 중단 후 개선되었다. 루푸스성 신장염이나 중추신경계 증상이 나타난 환자는 없었다.
9) 건선 : 발현 및 악화
이 약을 포함하여 TNF 저해제를 사용 시, 농포성 건선 및 손발바닥 건선을 포함하여 건선이 첫 발현되는 경우와 이미 존재하던 건선이 악화되는 경우들이 보고되었다. 이 환자들의 대다수는 면역억제제(예. MTX, 코르티코스테로이드)를 병용하고 있었으며, 일부는 입원치료가 필요했다. 대부분의 환자는 TNF 저해제의 복용을 중단함으로써 건선이 호전되었다. 일부 환자들은 이후, 다른 TNF 저해제를 재 투여했을 때 건선이 재발하였다. 국소 치료에도 불구하고 증상이 호전되지 않거나, 악화되는 심각한 경우에는 이 약의 투여 중지를 고려해야만 한다.
10) 간 효소 상승
◦ 류마티스 관절염 임상시험 : 류마티스 관절염에 대한 대조 임상시험에서(4건의 임상시험), ALT의 상승은 이 약 또는 위약을 투여한 환자에서 유사했다. 초기 류마티스 관절염 환자에서(3년 미만의 유병기간)(1건의 임상시험), ALT의 상승은 병용 투여군(이 약/메토트렉세이트)에서 메토트렉세이트 단독군 또는 이 약 단독군에 비해 더 자주 발생했다. 다관절형 소아 특발성 관절염 환자를 대상으로 한 임상시험에서 일부 트랜스아미나제의 상승이 나타났으나 위약과 이 약 투여군에서 같거나 유사했으며, 대부분은 메토트렉세이트와 병용 투여 시 나타났다.
◦ 건선성 관절염 임상시험 : ALT의 상승은 류마티스 관절염 임상시험의 환자에 비해 건선성 관절염 환자에 대한 임상시험(2건의 임상시험)에서 더 자주 발생하였다.
◦ 모든 류마티스 관절염, 다관절형 소아 특발성 관절염 및 건선성 관절염 임상시험에서, 상승된 ALT를 나타내는 환자들은 증상이 없었고, 대부분 상승이 일시적이며 투여 지속시 소실되었다.
◦ 크론병 및 궤양성 대장염 임상시험 : 대조 임상시험에서 이 약 투여군과 대조군간의 ALT상승은 유사하였다.
체중에 따라 조정된 유도 요법 후 체중에 따라 조정된 유지 요법(최대 52주)의 유효성과 안전성을 평가한 소아 크론병의 3상 임상시험에서, 시험대상자의 2.6%(5/192)에서 ≧ 3 X ULN의 ALT 상승이 발생하였고, 이 중 4명은 베이스라인에서 면역억제제를 병용 투여하였다.
◦ 건선 임상시험 : 건선에 대한 대조 임상시험에서, 투여군과 대조군간의 ALT 상승은 유사하였다.
◦ 화농성 한선염 임상시험 : 화농선 한선염에 대한 대조 임상시험에서, 투여군의 0.3%와 대조군의 0.6%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다.
◦ 소아 특발성 관절염 임상시험 : 4 ‑ 17세 다관절형 소아 특발성 관절염 환자와 6 ‑ 17세 골부착부위염 관절염 환자의 3상 대조 임상 시험에서, 투여군의 6.1%와 대조군의 1.3%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다. 대부분의 ALT 상승은 메토트렉세이트 병용 사용에 따라 발생하였다. 2세에서 4세 미만의 다관절형 소아 특발성 관절염 환자에 대한 3상 임상시험에서, ≧ 3 X ULN의 ALT 상승은 발생하지 않았다.
◦ 판상 건선 임상시험 : 판상 건선 소아 환자에 대한 3상 임상시험에서 ≧ 3 X ULN의 ALT 상승은 발생하지 않았다.
◦ 포도막염 임상시험: 포도막염에 대한 대조 임상시험에서, 투여군의 2.4%와 대조군의 2.4%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다.
모든 적응증의 임상시험에서 상승된 ALT를 나타내는 환자들은 증상이 없었고, 대부분 상승이 일시적이며 투여 지속 시 소실되었다. 그러나 간부전을 포함한 중증의 간손상이 이 약을 포함한 TNF 저해제를 투여받은 환자의 시판 후 보고에서 아주 드물게 나타났다. 이 약 투여와의 관계는 명백하지 않다.
11) 아자티오프린 또는 6 ‑메르캅토푸린과의 병용 투여 : 성인 크론병 임상시험에서, 아자티오프린/6 ‑메르캅토푸린과 병용 투여 시 이 약의 단독 투여에 비해 악성 종양 및 심각한 감염과 관련된 이상사례의 발생률이 더 높게 나타났다.
12) 시판 후 조사 또는 4상 시험에서 보고된 추가 이상사례 : 4상 시험과 시판 후 조사에서 보고된 추가 이상사례는 다음 표 2와 같다.
<표 2. 시판 후 조사 및 제4상 시험에서의 이상사례>
|
신체기관 |
이상사례 |
|
감염 및 기생충 감염 |
게실염 |
|
간담도계 이상* |
B형 간염의 재활성화, 간부전, 간염, 자가 면역성 간염 |
|
신경계 이상* |
탈수초성 장애(예. 시신경염), 길랑-바레 증후군, 뇌혈관사고 |
|
호흡, 흉부 및 종격계 이상 |
폐색전증, 흉막삼출, 폐섬유증 |
|
피부 및 피하 조직 이상 |
피부 혈관염, 스티븐스-존슨 증후군, 혈관부종, 건선의 발생 또는 악화(손발바닥 농포성 건선 포함), 다형홍반, 탈모, 태선모양 피부반응** |
|
면역계 이상* |
아나필락시스, 사르코이드증 |
|
위장관계 이상* |
장 천공 |
|
양성, 악성 및 미확인 신생물* |
Hepatosplenic T-cell lymphoma, 백혈병, 메르켈세포암(피부 신경내분비 선암) |
|
근골격계 및 결합조직 이상 |
루푸스 유사 증후군 |
|
심장 이상 |
심근경색증 |
|
전신 및 투여부위 이상 |
발열 |
|
* 1. 경고, 4. 일반적 주의 항에서 자세한 내용을 확인할 수 있다. ** 이 약을 포함한 TNF 저해제를 투여 받고 있는 환자들에게서 발생하였다. | |
⓵ 국내에서 6년 동안 류마티스 관절염, 건선성 관절염, 강직성 척추염, 크론병, 건선환자를 대상으로 실시한 시판 후 조사
국내에서 1,698명(류마티스 관절염 652명, 건선성 관절염 47명, 강직성 척추염 925명, 크론병 73명, 건선 1명)을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 10.07%(171명/1,698명, 238건)이었고, 이 중 이 약과 인과관계를 배제할 수 없는 약물이상반응 발현율은 6.83%(116명/1,698명, 141건)이며, 주사부위 반응 19건(1.11%), 가려움(증) 9건(0.53%), 발진 8건(0.47%), 대상포진, 상기도 감염, 두드러기 각 4건(0.24%), 신우신염, 홍반, 복통, 어지러움, 두통 각 3건(0.18%), 연조직염, 폐결핵, 결핵성 복막염, 폐렴, 결핵, 피부반응, 알레르기성 피부염, 림프종, 근육통, 기침, 림프절병증 각 2건(0.12%), 모낭염, 신장결핵, 비염, 편도염, 말라리아, 기관지 폐렴, B형 간염, 코 인두염, 감염성 윤활낭염, 진균성 감염, 크립토코쿠스 폐렴, 농포성 발진, 가려움 발진, 피부염, 탈모(증), 피부병변, 무력(증), 구토, 설사, 복막염, 구역, 고창, 갑상선암, 기관지 신생물, 급성 골수성 백혈병, 골관절염, 관절종창, 폐출혈, 콧물, 간손상, 간독성, 간기능 이상, 찔림 알레르기, 현기증, 월경 장애, 포도막염 각 1건(0.06%)이 보고되었다.
중대한 이상사례 발현율은 2.77%(47명/1,698명, 63건)이며, 신우신염, 복통이 각 4건이었으며 폐렴, 폐 결핵, 대상포진, 결핵성 복막염, 결핵, 설사, 구토, 가려움(증)이 각각 2건이며 그 외 신장결핵, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 항문 농양, 복막 농양, 결장염, 장관 천공, 복막염, 장관 경색, 장피부누공, 구역, 홍반, 발진, 대뇌출혈, 마비, 치매, 골관절염, 강직성 척추염, 등 통증, 갑상선암, 급성 골수성 백혈병, 직장암, 무력(증), 발열, 통증, 추골 탈구, 골절, 폐 출혈, 호흡곤란, 무릎 수술, 소장 절제(술), 혈중 크레아티닌 증가, 혈색소 감소, 간독성, 찔림 알레르기, 심근경색증, 빈혈, 탈수는 각 1건이 보고되었다.
이 중 이 약과 인과관계를 배제할 수 없는 중대한 약물이상반응 발현율은 1.47%(25명/1,698명, 30건)이며, 신우신염 3건, 복통, 폐결핵, 대상포진, 결핵성 복막염, 결핵, 가려움(증) 각 2건, 폐렴, 신장결핵, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 복막염, 홍반, 발진, 골관절염, 갑상선암, 급성 골수성 백혈병, 폐출혈, 혈중 크레아티닌 증가, 간독성, 찔림 알레르기 각 1건이 보고되었다.
예상하지 못한 이상사례 발현율은 3.24%(55명/1,698명, 61건)이며, 설사, 콧물 각 5건, 관절 종창, 포도막염 각 3건, 등 통증, 사지 통증, 감각 이상, 무력(증), 림프절병증 각 2건, 결장염, 복막염, 장관 경색, 위장관 궤양, 장피부누공, 명치불쾌감, 치루, 고창, 모낭염, 비염, 편도염, 말라리아, 기관지 폐렴, 감염성 윤활낭염, 크립토코쿠스 폐렴, 농포성 발진, 항문 농양, 복막 농양, 경부 통증, 강직성 척추염, 대뇌출혈, 마비, 치매, 신경근병증, 기억장애, 폐 출혈, 눈마름, 압통, 종양 절제(술), 간독성, 추골 탈구, 현기증, 피부병변은 각각 1건이 보고되었다.
이 중 이 약과 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 1.24%(21명/1,698명, 21건)이며, 림프절병증은 2건, 설사, 복막염, 고창, 모낭염, 비염, 편도염, 말라리아, 기관지 폐렴, 감염성 윤활낭염, 크립토코쿠스 폐렴, 농포성 발진, 관절 종창, 콧물, 폐 출혈, 포도막염, 무력(증), 간독성, 현기증, 피부병변은 각각 1건이 보고되었다. 중대하고 예상하지 못한 이상사례 발현율은 1.12%(19명/1,698명, 20건)이며, 설사는 2건, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 항문 농양, 복막 농양, 결장염, 복막염, 장관 경색, 장피부누공, 대뇌출혈, 마비, 치매, 강직성 척추염, 등 통증, 무력(증), 추골 탈구, 폐 출혈, 간독성은 각 1건이 보고되었고, 중대하고 예상하지 못한 약물이상반응 발현율은 0.35%(6명/1,698명, 6건)이며, 복막염, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 폐 출혈, 간독성 각 1건이 보고되었다.
⓶ 국내에서 4년 동안 소아 특발성 관절염(다관절형 소아 특발성 관절염, 골부착부위염 관련 관절염) 환자 28명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 21.43%(6명/28명, 8건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 17.86% (5명/28명, 7건) 이며, 독감 10.71% (3명/28명, 3건) 등통증, 관절부기, 두드러기, 발열 각 3.57% (1명/28명, 1건)이 보고되었다. 중대한 이상사례 발현율은 3.57%(1명/28명, 1건)이며, 홍반이 3.57% (1명/28명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응은 확인되지 않았다.
예상하지 못한 이상사례 발현율은 3.57% (1명/28명, 1건)이었고, 이 1건은 관절부기였으며 이는 본 제와 인과관계를 배제할 수 없다. 중대하고 예상하지 못한 이상사례 및 중대하고 예상하지 못한 약물이상반응은 확인되지 않았다.
⓷ 국내에서 4년 동안 소아 크론병 환자 143명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 18.18%(26명/143명, 47건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 14.69%(21명/143명, 26건)이며, 백혈구감소증 2.80%(4명/143명, 4건), 발진 2.10%(3명/143명, 3건), 설사, ALT증가, AST증가 각 1.40%(2명/143명, 2건), 대변내혈액, 마비성장폐쇄, 장천공, 가려움증, 국소피부반응, 두드러기, 상세불명의간기능검사이상, 충수돌기염, 권태, 주사부위홍반, 칸디다증, 신우신염, 모낭염이 각 0.70%(1명/143명, 1건) 보고되었다.
중대한 이상사례 발현율은 5.59%(8명/143명, 13건)이며, 복통 1.40%(2명/143명, 2건), 창자괴사, 대변내혈액, 마비성장폐쇄, 장폐쇄, 장천공, 소장폐쇄, 충수돌기염, 복막염, 열, 칸디다증, 신우신염이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응 발현율은 3.50%(5명/143명, 5건)이며, 마비성장폐쇄, 장천공, 충수돌기염, 칸디다증, 신우신염이 각 0.70%(1명/143명, 1건) 보고되었다.
예상하지 못한 이상사례 발현율은 6.29%(9명/143명, 17건)이며, AST증가 2.10%(3명/143명, 3건), 창자협착증 1.40%(2명/143명, 2건), 대장혈종, 위장염, 창자괴사, 마비성장폐쇄, 장폐쇄, 소장폐쇄, 충수돌기염, 헬리코박터파이로리위염, C반응단백질증가, 적혈구침강속도증가, 난치성통증, 명시안된수술후합병증이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 2.80%(4명/143명, 4건)이며, AST증가 1.40%(2명/143명, 2건), 마비성장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다.
중대하고 예상하지 못한 이상사례 발현율은 3.50%(5명/143명, 5건)이며, 창자괴사, 마비성장폐쇄, 장폐쇄, 소장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대하고 예상하지 못한 약물이상반응 발현율은 1.40%(2명/143명, 2건), 마비성장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다.
⓸ 국내에서 4년 동안 성인 화농성 한선염 환자 및 소아 판상 건선 환자 19명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 42.11%(8명/19명, 12건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 31.58%(6명/19명, 10건)이며, 한선염 15.79%(3명/19명, 4건), 여드름 5.26%(1명/19명, 2건), 소양증, 종기, 편도염, 발열이 각각 5.26%(1명/19명, 1건)으로 보고되었다. 중대한 이상사례 및 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응은 확인되지 않았다. 예상하지 못한 이상사례 발현율은 21.05%(4명/19명, 6건)이었고, 한선염 15.79%(3명/19명, 4건), 여드름 5.26%(1명/19명, 2건)으로 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 21.05%(4명/19명, 6건)이었다. 중대하고 예상하지 못한 이상사례 및 중대하고 예상하지 못한 약물이상반응은 확인되지 않았다.
⓹ 국내에서 포도막염에 대해 재심사를 위하여 4년 동안 155명을 대상으로 실시한 시판 후 조사 결과, 이상사례의 발현율은 인과관계와 상관없이 8.39%(13명/155명, 총 25건)로 보고되었다. 이 중 이 약과 인과관계를 배제할 수 없는 약물이상반응 발현율은 3.23%(5명/155명, 6건)이며, 안구 불편감, 주사부위 과민증, 주사 부위 통증, 지각 이상, 근육통, 습진 각 0.65%(1명/155명, 1건) 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응은 없었고, 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 1.29%(2명/155명, 2건) 안구 불편감, 주사부위 과민증 각 0.65%(1명/155명, 1건) 보고되었다.
➅ 이 약에 대한 국내 재심사 이상사례 및 자발적 부작용 보고자료를 국내 시판 허가된 모든 의약품을 대상으로 보고된 이상사례 보고자료(1989 ‑2021.5.31)와 재심사 종료시점에서 통합평가한 결과, 다른 모든 의약품에서 보고된 이상사례에 비해 이 약에서 통계적으로 유의하게 많이 보고된 이상사례 중 새로 확인된 것들은 다음과 같다. 다만, 이 결과가 해당성분과 다음의 이상사례 간에 인과관계를 입증된 것을 의미하는 것은 아니다.
◦ 근육 ‑골격계 이상 : 관절통, 관절병증
◦ 대사 및 영양계 이상 : 체중증가, 고지혈증
◦ 전신 및 투여부위 이상 : C반응단백질증가, 주사부위 덩어리, 주사부위 멍듦, 상태악화
◦ 호흡기계 이상 : 가래증가
◦ 정신계 이상 : 식욕증가
◦ 중추 및 말초신경계 이상 : 보행이상
◦ 생식기계 이상(여성) : 월경과다
◦ 소화기계 이상 : 흑색변
◦ 감염 : 농양, 종기증
◦ 피부 및 피하조직계 이상 : 각화과다증
14) 국내 관찰연구 결과
국내에서 베체트장염에 대해 6년 동안 50명을 대상으로 관찰 연구한 결과, 이상사례의 발현율은 인과관계와 상관없이 72%(36/50, 총 122건)로 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응 및 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 발현 빈도에 따라 아래 표에 나열하였다.
|
|
중대한 약물이상반응 8%(4명/50명, 11건) |
예상하지 못한 약물이상반응 14%(7명/50명, 9건) | |
|
때때로(0.1~5%미만) |
각종 위장관 장애 |
복통, 상복부 통증, 설사, 오심 |
입 궤양 형성, 베체트 증후군, 상복부 통증, 변비 |
|
감염 및 기생충 감염 |
급성 신우신염, 파종 결핵, 인두염 |
| |
|
각종 정신 장애 |
|
수면 장애 | |
|
전신 장애 및 투여 부위 병태 |
발열 |
주사 부위 경화 | |
|
대사 및 영양 장애 |
섭식 저하 |
섭식 저하 | |
|
호흡기, 흉곽 및 종격 장애 |
기침 |
| |
|
피부 및 피하 조직 장애 |
|
접촉 피부염 | |
1) 알레르기 반응 : 임상시험 동안 이 약의 피하주사로 인한 심각한 알레르기 반응은 드물게 보고되었다. 이 약 투여 후 아나필락시스를 포함한 중대한 알레르기 반응이 보고된 바 있다. 만일 아나필락시스 반응이나 다른 심각한 알레르기 반응이 나타나면 이 약의 투여를 즉시 중단하고 적절한 치료를 시작해야 한다.
2) 면역억제 : 이 약을 투여받은 64명의 류마티스 관절염 환자에 대한 시험에서 지연성 과민반응의 억제, 면역글로불린 억제 또는 effector T ‑와 B세포, NK ‑세포, 단핵세포/대식세포 및 호중구의 수적 변화의 증거는 없었다.
3) 혈액학적 반응 : TNF 저해제에서 드물게 재생불량성빈혈을 포함한 범혈구감소증이 보고되었다. 의학적으로 중요한 혈구감소증(예. 저혈소판혈증, 백혈구감소증)을 포함한 혈액계의 이상사례가 이 약에서 보고되었다. 이러한 보고의 이 약에 대한 원인 상관관계는 불분명하다. 이 약을 투여하는 동안 혈액질환을 암시하는 징후 또는 증상(예, 지속적인 발열, 타박상, 출혈, 창백)이 발현되는 경우 즉각적인 의학적 처치를 받도록 모든 환자를 교육해야 한다. 심각한 혈액학적 이상이 확정된 환자는 이 약의 투여 중단을 고려해야 한다.
4) 예방접종 : 이 약 또는 위약을 투여한 226명의 성인 류마티스 관절염 환자에 대한 시험에서 표준 23가 폐구균 백신과 3가 인플루엔자 백신에 대한 항체반응과 유사한 반응이 관찰되었다. 이 약을 투여받은 환자에게 생백신을 투여한 경우 이에 의한 이차적인 감염 전파에 대한 이용 가능한 자료는 없다. 이 약을 투여 받고 있는 환자에게 생백신을 제외한 다른 백신을 병용 투여할 수 있다.
5) 울혈성 심부전 : 다른 TNF 저해제를 사용한 임상시험에서 울혈성 심부전의 악화와 울혈성 심부전으로 인한 사망률 증가가 관찰되었다. 이 약을 투여받은 환자에서도 울혈성 심부전이 악화된 사례가 보고되었다. 경증 심부전(NYHA classI/II) 환자에게 이 약 투여시 주의해야 하며 중등도 또는 중증 심부전 환자에게 투여해서는 안 된다. 울혈성 심부전이 새롭게 발생하거나 증상이 악화된 환자의 경우 이 약의 투여를 중단해야 한다.
6) 자가면역 과정 : 이 약의 투여로 자가면역 항체가 형성될 수 있다. 이 약의 장기투여가 자가면역질환 발생에 미치는 영향은 알려지지 않았다. 만일 이 약으로 치료 후 환자에게서 루푸스양 징후를 암시하는 증상을 나타내고 dsDNA에 대한 항체가 양성이라면 이 약의 투여를 중지해야 한다.
7) 생물학적 DMARDs 또는 TNF 저해제와의 병용투여 : 에타너셉트(다른 TNF 저해제)와 아나킨라를 병용 투여한 임상 시험에서 심각한 감염이 관찰되었으며, 에타너셉트 단독 요법에 비해 추가적인 유익성이 없었다. 에타너셉트와 아나킨라의 병용 시 관찰된 이상사례의 특성 때문에, 아나킨라와 다른 TNF 저해제와의 병용으로 인해 유사한 독성이 있을 수 있다. 이 약과 아나킨라의 병용 투여가 권장되지 않는다. 감염 및 기타 잠재적 약물학적 상호작용의 위험 증가 가능성으로 이 약과 다른 생물학적 DMARDs (예. 아나킨라 및 아바타셉트) 또는 다른 TNF 저해제와의 병용투여는 권장되지 않는다.
8) 수술 : 이 약을 투여받은 환자에서 수술 절차의 안전성 경험은 제한적이다. 만일 수술 절차가 계획된다면 이 약의 긴 반감기를 고려해야 한다. 이 약 투여동안 수술이 필요한 환자는 감염에 대해 면밀히 모니터링하고 적절한 조치를 취해야 한다. 이 약 투여 동안 관절성형술을 받는 환자에 대한 안전성 경험은 제한적이다.
9) 소장 폐색증 : 크론병의 치료에 반응하지 않은 환자는 외과적 치료를 요하는 고정된 섬유성 협착을 보일 수 있다. 이 약이 협착을 일으키거나 악화시키지 않는다는 것을 나타내는 자료가 있다.
10) 간 ◦신장애 환자 : 이러한 환자군에 대해서 연구되지 않았으며 권장 용량이 없다.
11) 이 약이 운전이나 기계 조작 능력에 미치는 영향에 대해서는 연구된 바 없다.
5. 상호작용
1) 이 약은 류마티스 관절염 환자에서 단독요법 및 메토트렉세이트와의 병용 요법 모두에 대해 연구되었다. 시험결과 이 약 또는 메토트렉세이트의 용량조절 필요성은 나타나지 않았다. 공식적인 약동학 시험에서 이 약과 메토트렉세이트 이외의 약물과의 상호작용을 평가하지 않았다. 임상시험에서 이 약과 일반적으로 사용되는 DMARDs(sulfasalazine, hydrochloroquine, leflunomide 및 parenteral gold), 글루코코르티코이드, 살리실산염, 비스테로이드성항염증제 또는 진통제를 병용 투여 시 상호작용이 나타나지 않았다.
이 약 단독투여에 비해 메토트렉세이트와 병용 투여 시 항체 생성률이 더 낮았다. 메토트렉세이트를 투여하지 않고 이 약 단독투여시 항체 생성률과 이 약의 제거율이 증가했으며, 이 약의 효과는 감소했다.
2) 생백신과 이 약의 병용 투여는 권장되지 않는다.
3) 아나킨라와 이 약의 병용 투여는 권장되지 않는다.
4) 약물/실험실 검사 상호작용 : 이 약과 실험실 검사 사이에 알려진 간섭은 없다.
6. 임부 및 수유부에 대한 투여
1) 원숭이에 대한 발생독성시험에서 모체 독성, 배아 독성 또는 최기형성이 나타나지 않았다. 출생후 독성과 수태능에 미치는 영향에 대한 비임상시험 자료는 없다. TNF 알파 저해 작용으로 인해 임신기간 중 이 약 투여는 신생아의 정상 면역반응에 영향을 줄 수 있다. 임신기간 동안 이 약은 태아에 대한 잠재적 유익성이 잠재적 위험성을 상회하는 경우에만 임부에게 투여해야한다. 임신할 가능성이 있는 여성은 임신을 예방하기 위한 적절한 피임요법 사용과 이 약의 최종 투여 후 최소 5개월간 피임을 지속할 것을 고려하여야 한다.
임신 중 노출에 대한 전향적 코호트 등록 연구에서, 임신 1기 동안 아달리무맙을 최소 1 회 투여한 류마티스 관절염 또는 크론병이 있는 257명의 여성과 아달리무맙을 투여받지않은 류마티스 관절염 또는 크론병이 있는 120명의 여성이 등록되었다.
일차 평가변수는 주요출생결함의 발생률이었다. 주요 출생결함이 있는 한 명이상의 생존 신생아 출산으로 종료된 임신율은 아달리무맙을 투여받은 여성에서 6/69(8.7%), 치료를 받지 않은 류마티스관절염 여성에서 5/74 (6.8%) (보정하지 않은 odd ratio 1.31, 95% 신뢰구간 0.38 ‑4.52)이었고, 아달리무맙을 투여받은 크론병 16/152 (10.5%), 치료를 받지 않은 크론병 여성에서 3/32 (9.4%)(보정하지 않은 odd ratio 1.14, 95% 신뢰구간 0.31 ‑4.16)이었다. 류마티스관절염과 크론병의 베이스라인 차이를 보정한 odd ratio는 1.10 (95% 신뢰구간 0.45 ‑2.73)이었다.
이차 평가변수인 경미한 출생결함, 조기분만, 저체중, 심각한 또는 기회 감염에서 유의한 차이가 관찰되지 않았다 (보정한 odd ratio 0.84, 95% 신뢰구간 0.34, 2.05). 사산이나 악성 종양은 보고되지 않았다.
이 등록연구는 적은 표본 크기와 무작위연구 설계가 아니라는 방법론적인 한계로 데이터 해석에 영향을 줄 수 있다.
아달리무맙은 임신 중 아달리무맙으로 치료받은 여성에서 태반을 통과하여 태아의 혈청으로 들어간다. 결과적으로 이 유아들은 감염의 위험이 증가될 수 있다. 자궁에서 아달리무맙에 노출된 유아의 경우, 임신 중 이 약의 마지막 투여 후 5개월 동안 생백신 투여는 권장되지 않는다.
2) 출판된 문헌의 제한된 정보에서 모유로 분비되는 아달리무맙의 농도는 매우 낮으며, 인간의 모유 내 아달리무맙 농도는 모체 혈청 수치의 0.1%에서1% 정도이다. 경구로 섭취한 면역글로불린 G 단백질은 장내에서 단백질 분해되고 생체이용률이 낮으므로, 모유수유하는소아에 대한 아달리무맙의 전신 영향 가능성은 거의 없을 것이다. 모유수유의 발달 및 건강상 이익은 아달리무맙에 대한 수유부의 임상적 필요성과 아달리무맙 또는 수유부의 기존 상태로 인한 소아에 대한 잠재적인 유해성과 함께 고려해야 한다.
7. 소아에 대한 투여
이 약은 소아 환자에 대해 연구되지 않았으므로 추가적인 자료를 얻을 때까지 18세 미만 소아에 대한 투여는 권장되지 않는다[소아 특발성 관절염(다관절형 소아 특발성 관절염, 골부착부위염 관련 관절염), 크론병(6 ‑ 17세) 및 판상 건선 제외].
8. 고령자에 대한 투여
이 약의 용량 조절이 요구되지 않는다. 이 약의 임상시험에 참가한 환자 중 65세 이상이 9.4%이고 75세 이상이 약 2.0%였으며 이들 환자군과 더 젊은 환자군 사이에 전반적인 유효성의 차이는 관찰되지 않았다. 이 환자군에서 용량조절은 필요하지 않다.
9. 과량투여시의 처치
임상시험 중 용량제한독성은 관찰되지 않았다. 평가된 최고 용량은 10 mg/kg의 수회 정맥투여였다. 과량투여의 경우 이상사례의 증상이나 징후 또는 효과를 모니터링하고 적절한 대증적 처치를 한다.
10. 적용상의 주의사항
1) 주사방법에 대한 교육을 받은 후 환자가 자가주사할 경우, 자가주사 부위는 대퇴부 또는 복부를 포함하며 주사부위는 교대로 바꿔야 한다. 새로운 주사부위는 앞서 주사한 부위에서 최소 3 cm 정도 떨어져서 주사한다.
2) 새로운 주사부위가 약하거나, 멍들거나, 충혈되거나 딱딱한 경우에는 투여해서는 안 된다. 투여 전 변색 및 이물질에 대해 육안으로 관찰한다. 주사액은 무색투명해야 하며, 주사액이 변색 또는 불투명하거나 이물이 있을 경우 투여하지 않는다. 펜형 제품의 경우 몸체에 있는 창(window)을 통해 확인한다. 주사 전 제품을 흔들거나 떨어뜨리지 않는다.
회색 뚜껑 및 자주색 뚜껑을 주사 직전에 제거한다.
3) 프리필드시린지
주사부위를 동봉된 알콜솜으로 닦는다. 주사바늘 덮개를 벗기고, 주사부위를 부드럽게 잡아 고정한 후 피부와 45도 각도로 주사한다.
4) 펜(pen) 형
⓵ 주사부위를 동봉된 알콤솜으로 닦는다. 회색 뚜껑 및 자주색 뚜껑을 주사 직전에 제거한다. 펜의 회색부분을 한손으로 잡는다. 펜의 회색 뚜껑(1) 및 자주색 뚜껑(2)에 손이 닿지 않도록 펜의 가운데를 손으로 잡는다. 회색 뚜껑(1)이 위로 향하도록 잡는다.
다른 손으로 회색 뚜껑(1)을 즉시 당겨서 제거한다. 작은 회색 주사바늘 커버와 덮개가 함께 제거되었는지 확인하며, 뚜껑을 다시 씌우면 안 된다. 주사침에서 몇 방울의 액이 나오는 것은 정상이다.
⓶ 펜의 자주색 뚜껑(2)을 잡아당겨 벗긴 후 버린다. 자주색 작동 버튼이 노출되어 펜을 사용할 준비가 된다. 약물이 방출될 수 있으므로, 적절한 주사위치를 잡기 전까지 자주색 작동 버튼을 누르거나 뚜껑을 다시 씌우면 안 된다.
⓷ 한 손으로 주사부위의 깨끗한 피부를 아래 그림처럼 부드럽게 잡아 고정시킨다.
⓸ 펜의 흰색 끝부분을 창이 보이도록 하여 피부와 직각(90도)이 되도록 놓는다. 창으로 보여 지는 몇 방울의 기포는 정상이다.
⓹ 펜의 몸체를 잡고 주사부위에 가볍게 눌러 주사준비를 한 다음 집게손가락 또는 엄지손가락으로 주사기 윗부분의 자주색 버튼을 누른다. 바늘이 나올 때 나는 ‘딸깍’ 소리가 들리고, 바늘에 찔리는 따끔함이 느껴진다.
➅ 약 10초간 유지하여 완전히 주사되도록 한다.
⑦ 주사 동안 노란색 표시가 펜의 창내로 이동하며, 노란색 표시의 이동이 멈추면 주사가 끝난 것이다. 노란색 표시는 펜의 주사막대의 일부분이다. 노란색 표시가 창에 나타나지 않는다면, 주사막대가 적절히 이동하지 않은 것이고, 주사가 완전히 이루어지지 않은 것이다.

5) 이 약의 주사기에 다른 약물을 혼합해서는 안 된다. 사용하지 않고 남은 부분은 버린다. 일회용이며 재사용하지 않는다. 어린이의 손이 닿지 않는 곳에 보관한다.
11. 저장상의 주의사항
1) 이 약은 동결을 피하여 냉장 보관(2 ‑ 8℃)한다. 사용 전까지 포장을 뜯지 않고 용기 상자 내부에 보관한다.
2) 이 약은 냉장조건을 벗어난 경우 1회에 한하여 25℃이하에서 최대 14일간 보관할 수 있으며, 빛을 피하여 보관하여야 한다. 냉장조건을 벗어난 제품은 다시 냉장보관해서는 안되며, 14일 이내 사용하거나 폐기하여야 한다.
12. 기타
이 약은 류마티스 관절염 환자에 대한 한 건의 이중 눈가림 후 공개연장 임상시험에서 최대 60개월까지 투여되었다. 60개월까지의 투여기간 동안 류마티스 관절염의 증상과 징후를 감소시키는 효과가 유지되었고 관절손상 진행속도 감소와 신체활동기능 향상 효과가 유지되었으며, 전반적으로 안전하고 내약성이 좋았다.
13. 전문가를 위한 정보
1) 임상약리학적 정보
⓵ 일반 정보
아달리무맙은 TNF에 특이적으로 결합하여 TNF와 p55 및 p75 세포 표면 TNF 수용체의 상호작용을 차단함으로써 TNF의 생물학적 기능을 중화시킨다. TNF는 자연적으로 발생하는 사이토카인으로서 정상적인 염증 및 면역 반응에 관여한다. 또한 아달리무맙은 백혈구 이동(ELAM ‑1, VCAM ‑1 및 ICAM ‑1, IC50 1 ‑2 × 10 ‑10M)의 원인이 되는 부착 분자의 수치 변화와 같이 TNF에 의해 유도되거나 조절되는 생물학적 반응을 조절한다.
⓶ 약력학적 정보
이 약을 투여한 후 류마티스 관절염 환자에서 베이스라인과 비교하여 염증의 급성 단계 반응물질(C ‑반응성 단백질[C ‑reactive protein; CRP]과 적혈구 침강속도[erythrocyte sedimentation rate; ESR] 과 혈중 사이토카인[IL ‑6]) 수치가 급격히 감소하였다. 또한 소아 특발성 관절염, 크론병, 궤양성 대장염 및 화농성 한선염 환자에서 CRP 수치 감소뿐만 아니라 크론병 환자의 대장에서 인간 백혈구 항원(human leukocyte antigen; HLA ‑DR) 및 골수세포형과산화효소(myeloperoxidase; MPO)와 같은 염증 표지자와 TNF가 유의하게 감소하였다. 연골 손상의 원인이 되는 조직 리모델링을 일으키는matrix metalloproteinase(MMP ‑1 및 MMP ‑3)의 혈중 수치 또한 이 약의 투여 후 감소하였다. 류마티스 관절염, 건선성 관절염, 강직성 척추염 환자는 경증부터 중등도의 빈혈 및 림프구 수 감소뿐 아니라 호중구와 혈소판 수 증가를 종종 경험한다. 이 약을 투여 받은 환자는 대개 만성 염증의 혈액학적 징후에서 개선을 경험하였다.
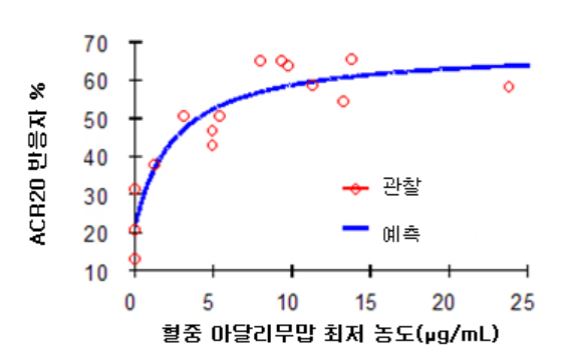
그림 1: 농도 ‑효과 연관성
⓷ 약동학적 정보
흡수
건강한 성인 대상자 59명에게 아달리무맙 40 mg을 단회 피하 투여한 아달리무맙의 흡수 및 분포는 느리며 투여 후 약 5일 뒤에 평균 최대 혈중 농도에 도달하였다. 3건의 시험에서 40 mg 단회 피하 투여 후 추정한 아달리무맙의 절대 생체이용률은 평균 64%였다.
분포 및 배설
아달리무맙의 단일 투여 약동학은 0.25 ‑10 mg/kg 범위의 정맥 내 투여에 대한 여러 시험에서 확인되었다. 4.7 ‑6.0 L 범위의 분포 용적(distribution volume; Vss)은 아달리무맙이 혈관과 혈관 외액 사이에 거의 비슷하게 분포함을 나타내었다. 아달리무맙은 일반적으로 12 mL/h 이하의 청소율로 천천히 배설된다. 평균 최종 단계 반감기는 약 2주였으며 범위는 10 ‑20일이었다. 청소율 및 반감기는 시험된 투여량 범위에서 상대적으로 변화가 없었고 최종 반감기는 IV 투여 및 SC 투여에서 비슷하였다. 여러 류마티스 관절염환자 활액에서의 혈중 아달리무맙 농도 범위는 31 ‑96%였다.
정상상태(steady ‑state)에서의 약동학
류마티스 관절염 환자에게 격주로 아달리무맙 40 mg을 SC 투여한 후의 반감기를 근거로 아달리무맙의 축적을 예측할 수 있었으며 평균 정상상태 최저 농도는 각각 약 5 mcg/mL(메토트렉세이트[MTX] 병용 투여하지 않은 경우) 및 8 ‑9 mcg/mL (MTX 병용 투여한 경우)였다. 정상상태 혈중 아달리무맙 최저 수치는 격주 및 매주 SC 투여한 20, 40 및 80 mg의 투여량에 따라 거의 비례적으로 증가하였다. 2년 이상의 장기간 투여 시험에서 시간에 따른 청소율의 변화는 확인되지 않았다.
건선 환자의 경우 메토트렉세이트의 병용 투여 없이 아달리무맙 40 mg 격주 투여 동안 평균 정상상태 최저 농도는 5 mcg/mL였다.
0주차에 아달리무맙 160mg을 투여 받은 후 2주차에 80 mg을 투여 받은 화농성 한선염 환자의 혈중 아달리무맙 최저 농도는 2주차와 4주차에 약 7 ‑8 mcg/mL에 도달하였다. 12주차부터 36주차까지의 평균 정상상태 최저 농도는 매주 아달리무맙 40 mg 투여 동안 약 8 ‑10 mcg/mL였다.
포도막염 환자의 경우 0주차에 도입용량인 아달리무맙 80 mg을 투여하고 1주차부터 격주마다 아달리무맙 40 mg을 투여했을 때 평균 정상상태 농도는 약 8 ‑10 mcg/mL였다.
집단 약동학과 약동학/약력학 모델링 및 시뮬레이션 결과, 매주 40 mg의 아달리무맙 투여와 비교하여 격주 80 mg 투여한 환자의 아달리무맙 노출 및 효능이 유사한 것으로 예측하였다(성인 류마티스 관절염, 화농성 한선염, 궤양성 대장염, 크론병 또는 건선환자 및 40kg 이상의 소아 크론병 환자).
1,200명 이상 환자 데이터의 집단 약동학 분석을 통해 메토트렉세이트의 병용 투여가 아달리무맙의 겉보기 청소율(apparent clearance; CL/F)에 본질적인 영향을 미친다는 것이 확인되었다(‘5. 상호작용’ 참조). 예상한 바와 같이 체중 증가 및 항 ‑아달리무맙 항체의 존재 하에 아달리무맙의 겉보기 청소율이 높아지는 경향이 있었다.
보다 중요하지 않은 다른 요인들도 확인하였다. 권장된 투여량보다 낮은 투여량을 받은 환자, 높은 류마티스인자를 가진 환자 또는 CRP 농도가 높은 환자의 겉보기 청소율은 높을 것으로 예측되었다. 이러한 요인은 임상적으로 중요할 가능성이 낮다.
비방사선학적 축성 척추관절염 환자에게 격주마다 아달리무맙 40 mg을 피하투여한 후 68주차의 평균(±SD) 최저 정상상태 농도는 8.0 ± 4.6 μg/mL였다.
크론병 환자의 경우, 0주차에 유도용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 2주차와 4주차의 평균 혈중 아달리무맙 최저 수치는 약 12 mcg/mL였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 크론병 환자에서 24주차와 56주차의 평균 정상상태 최저 수치는 약 7 mcg/mL로 관찰되었다.
궤양성 대장염 환자의 경우, 0주차에 유도용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 유도 기간 동안의 혈중 아달리무맙 최저 농도는 약 12 mcg/mL에 도달하였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 궤양성 대장염 환자에서의 평균 정상상태 최저 수치는 약 8 mcg/mL로 관찰되었다.
베체트병이 있는 일본 환자의 경우, 0주차에 도입용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 유도 기간 동안의 혈중 아달리무맙 최저 농도는 약 13 mcg/mL에 도달하였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 장내 베체트병이 있는 일본 환자에서의 평균 정상상태 최저 수치는 약 9 mcg/mL로 관찰되었다.
특수 집단
특수한 집단에서의 약동학은 집단 약동학 분석을 사용하여 조사하였다.
고령 환자
연령은 아달리무맙의 겉보기 청소율에 대해 최소한의 영향을 미치는 것으로 나타났다. 집단 분석에서 40 ‑ 65세 환자(n=850) 및 ≧ 65세 환자(n=287)의 평균 체중을 보정한 청소율은 각각 0.33 및 0.30mL/h/kg이었다.
소아 환자
다관절형 소아 특발성 관절염이 있는 4 ‑ 17세의 환자에게 격주마다 아달리무맙 24 mg/m2(최대 40 mg)를 피하투여한 후 평균 최저 정상상태(20 ‑48주차에 측정한 수치) 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여했을 때 5.6 ± 5.6 μg/mL (102% CV), 메토트렉세이트를 병용 투여하지 않았을 때 10.9 ± 5.2 μg/mL (47.7% CV)였다. 20 mg 아달리무맙을 격주마다 피하투여한 30 kg인 환자에서 메토트렉세이트를 병용 투여하거나 병용 투여하지 않았을 때 평균 정상상태 혈중 아달리무맙 농도는 각각 6.8 μg/mL 및 10.9 μg/mL였다. 40 mg 아달리무맙을 격주마다 피하투여한 ≧30 kg인 환자에서 메토트렉세이트를 병용 투여하거나 병용 투여하지 않았을 때 평균 정상상태 혈중 아달리무맙 농도는 각각 6.6 μg/mL 및 8.1 μg/mL였다. 다관절형 소아 특발성 관절염이 있는 2세이상 4세미만 또는 ≧4세의 15 kg인 환자에게 아달리무맙 24 mg/m2를 투여했을 때의 평균 최저 정상상태 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여하지 않은 경우 6.0 ± 6.1 μg/mL (101% CV), 메토트렉세이트를 병용 투여한 경우 7.9 ± 5.6 μg/mL (71.2% CV)였다.
골부착부염 관련 관절염 환자에게 24 mg/m2(최대 40 mg)를 격주마다 피하투여한 후 평균 최저 정상상태(24주차에 측정한 수치) 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여하지 않은 경우 8.8 ± 6.6 μg/mL, 메토트렉세이트를 병용 투여한 경우 11.8 ± 4.3 μg/mL였다.
중등증부터 중증의 활동성 크론병 소아 환자에서의 공개 연장 시험 아달리무맙 유도용량은 0주차와 2주차에 체중 절단점(cut ‑off)인 40 kg에 따라 각각 160/80 mg 또는 80/40 mg이었다. 4주차에 환자는 체중에 따라 표준용량(격주 40/20 mg) 또는 저용량(격주 20/10 mg) 유지 투여군에 1:1의 비율로 무작위 배정되었다. 4주차에 도달한 평균(±SD) 혈중 아달리무맙 최저 농도는 ≧40 kg인 환자(160/80mg)의 경우 15.7 ± 6.6 μg/mL, 40 kg인 환자(80/40 mg)의 경우 10.6 ± 6.1 μg/mL였다.
무작위 배정된 치료를 지속한 환자에서 52주차의 평균(±SD) 아달리무맙 최저 농도는 표준용량 투여군의 경우 9.5 ± 5.6 μg/mL, 저용량 투여군의 경우 3.5 ± 2.2 μg/mL였다. 52주 동안 아달리무맙 격주 투여를 지속한 환자에서 평균 최저 농도는 유지되었다. 격주에서 매주 투여 요법으로 투여량을 증량한 환자의 경우 52주차의 평균(±SD) 아달리무맙 혈중 농도는 각각 15.3 ± 11.4 μg/mL(매주 40/20 mg) 및 6.7 ± 3.5 μg/mL(매주 20/10 mg)였다.
만성 판상형 건선이 있는 소아 환자에게 격주로 아달리무맙 0.8 mg/kg(최대 40 mg)을 피하투여한 후 평균 ±SD 정상상태 아달리무맙 최저 농도는 약 7.4 ± 5.8 μg/mL (79% CV)였다.
성별
환자의 체중을 보정한 후 성별과 관련된 약동학 차이는 관찰되지 않았다.
인종
인종간 면역글로불린 청소율의 차이는 예상되지 않는다. 코카시안이 아닌 인종의 제한된 데이터로부터, 아달리무맙에 대한 중요한 약동학적 차이는 관찰되지 않았다.
간장애 및 신장애 환자
간 또는 신장 기능 장애가 있는 환자에서 유효한 약동학 데이터가 없다.
질병 상태
건강한 지원자와 류마티스 관절염 환자는 유사한 아달리무맙 약동학을 보였다.
2) 임상시험 정보
⓵ 성인
류마티스 관절염
모든 류마티스 관절염 임상시험에서 3,000명 이상의 환자를 대상으로 이 약을 평가하였다. 이 약의 안전성 및 유효성은 5건의 무작위배정, 이중눈가림 된 시험에서 평가하였다. 일부 환자는 60개월 이상의 기간 동안 이 약을 투여 받았고, 일부는 최대 120개월의 기간 동안 이 약을 투여 받았다. 이 약 40 mg/0.8 mL과 비교한 이 약 40 mg/0.4 mL에 대한 투여 부위 통증은 2건의 무작위 배정, 활성대조, 단일 눈가림, 두 기간 교차시험으로 평가하였다.
RA 시험 I (DE009)은 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 271명을 대상으로 평가하였으며, 이 환자들은 1 ‑4개의 DMARDs(disease ‑modifying anti ‑rheumatic drug),(예: 히드록시클로로퀸, 경구 또는 주입 가능한 금, 아자티오프린, D ‑페니실라민, 설파살라진) 치료에 실패 이력이 있고 매주 12.5 ‑25 mg (메토트렉세이트 내약성이 없는 경우 10 mg)의 메토트렉세이트에 대해 효능이 불충분하며 메토트렉세이트 투여량을 매주 10 ‑25 mg으로 일정하게 유지하였다. 환자는 6개 이상의 종창 관절 및 9개 이상의 압통 관절이 있었으며 ACR 기준에 따라 류마티스관절염로 진단되었다. 이들은 24주 동안 격주마다 20, 40 또는 80 mg의 이 약이나 위약을 투여 받았다.
RA 시험 II (DE011)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 544명을 대상으로 평가하였으며, 이들은 최소 하나의 DMARD(예: 메토트렉세이트, 설파살라진, 히드록시클로로퀸, 경구 또는 주입 가능한 금, D ‑페니실라민, 아자티오프린) 치료에 실패한 이력이 있었다. 환자는 10개 이상의 종창 관절 및 12개 이상의 압통 관절이 있었으며 ACR 기준에 따라서도 진단되었다. 이들은 26주 동안 이 약 20 mg 또는 40 mg을 위약과 격주로 번갈아 피하 투여 받거나 매주 피하 투여 받았다. 위약은 동일한 기간 동안 매주 투여 받았다.
RA 시험 III (DE019)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 619명을 대상으로 평가하였으며, 매주 12.5 ‑25 mg (메토트렉세이트 내약성이 없는 경우 10 mg)의 메토트렉세이트에 대해 효능이 불충분하며 메토트렉세이트 투여량을 매주 12.5 ‑25 mg으로 일정하게 유지하였다. RA 시험 I과는 다르게, RA 시험 III의 환자는 메토트렉세이트 외의 DMARD 치료 실패 이력이 필요하지 않았다. 환자는 6개 이상의 종창 관절 및 9개 이상의 압통 관절이 있었으며 ACR 기준에 따라 류마티스 관절염으로 진단되었다. 이 시험에는 3개의 군이 있었다. 첫 번째 군은 52주 동안 매주 위약을 투여 받았다. 두 번째 군은 52주 동안 매주 이 약 20 mg 을 투여 받았다. 세 번째 군은 이 약 40 mg과 위약을 격주로 번갈아 투여 받았다. 첫 52주 동안 투여를 완료한 후 457명의 환자는 최대 60개월 동안 격주로 이 약 40 mg/메토트렉세이트를 투여 받는 공개 연장 시험에 등록되었다. 첫 52주 동안 투여를 완료한 후 457명의 환자는 최대 10년 동안 격주로 이 약 40mg /메토트렉세이트를 투여 받는 공개 연장 시험에 등록되었다.
RA 시험 IV (DE031)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 636명을 평가하였다. 이 환자들은 최소 3개월 동안 류마티스 관절염 진단에 대한 ACR 기준을 충족했으며 최소 6개의 종창 관절 및 9개의 압통 관절을 가지고 있었다. 최소한 28일 동안 치료가 안정적이었을 경우, DMARD ‑I 또는 기존의 류마티스 치료를 유지하는 것이 허용되었다. 환자는 24주 동안 이 약 격주 40 mg투여군 또는 위약군에 무작위 배정되었다.
RA 시험 V (DE013)는 중등증에서 중증의 활동성 초기 류마티스 관절염(평균 질병 기간이 9개월 미만)이 있는 메토트렉세이트 투여 이력이 없는 성인 환자 799명을 대상으로 평가하였다. 이 시험에서는 104주 동안 류마티스 관절염에서 류마티스 관절염의 징후 및 증상을 감소시키고 관절 손상의 진행 속도를 줄이는 데 대한 이 약 40 mg 격주/메토트렉세이트 병용 치료, 이 약 40 mg 격주 단일 치료 및 메토트렉세이트 단일 치료의 효능을 평가하였다. 첫 104주 동안 투여를 완료한 후 497명의 환자는 최대 10년 동안 이 약 격주로 40 mg을 투여 받는 공개 연장 시험에 등록되었다.
RA 시험 VI 및 VII는 각각 중등증에서 중증의 활동성 류마티스 관절염이 있는 ≧ 18세 환자 60명을 대상으로 평가하였다. 등록된 환자는 당시 이 약 40 mg/0.8 mL를 투여 받는 중이며 평균 투여 부위 통증이 최소 3cm (0 ‑10cm VAS)인 환자, 또는 이 약 40 mg/0.8 mL를 투여 받기 시작한 생물학적 치료 이력이 없는 환자였다. 환자는 이 약 40 mg/0.8 mL 또는 이 약 40 mg/0.4 mL 단일 투여군으로 무작위 배정된 후 다음 파트에 교차 투여된다.
RA 시험 I ‑V의 결과는 ACR 반응 기준을 사용하여 류마티스 관절염에서 개선을 보인 환자 비율로 표현하였다. RA 시험 I, II 및 III의 일차 평가변수와 RA 시험 IV의 이차 평가변수는 24주 또는 26주차에 ACR 20 반응에 도달한 환자의 비율이었다. RA 시험 V의 일차 평가변수는 52주차에 ACR 50 반응에 도달한 환자 비율이었다. RA 시험 III 및 V의 52주차에서 추가적인 일차 평가변수는 질병 진행의 지연이었다(엑스레이 결과로 확인). RA 시험 III 또한 삶의 질 변화를 일차 평가변수로 설정하였다. RA 시험 VI 및 VII의 일차 평가변수는 0 ‑10 cm VAS로 측정한 투여 직후의 투여 부위 통증이었다.
임상 반응
RA 시험 I, II 및 III
이 약을 투여 받은 환자 중 ACR 20, 50 및 70 반응에 도달한 환자 비율은 3건의 시험 모두에서 일관적이었다. 3건의 시험 결과는 표 3에 요약한다.
표 3: 위약대조 시험에서의 ACR 반응(환자 비율)
|
반응 |
RA 시험 Ia* |
RA 시험 IIa* |
RA 시험 IIIa* | |||
|
|
위약/MTX |
이 약b/MTX |
위약/MTX |
이 약b/MTX |
위약/MTX |
이 약b/MTX |
|
|
n=60 |
n=63 |
n=110 |
*n=113 |
n=200 |
n=207 |
|
ACR 20 |
|
|
|
|
|
|
|
6개월 |
13.3% |
65.1% |
19.1% |
46.0% |
29.5% |
63.3% |
|
12개월 |
NA |
NA |
NA |
NA |
24.0% |
58.9% |
|
ACR 50 |
|
|
|
|
|
|
|
6개월 |
6.7% |
52.4% |
8.2% |
22.1% |
9.5% |
39.1% |
|
12개월 |
NA |
NA |
NA |
NA |
9.5% |
41.5% |
|
ACR 70 |
|
|
|
|
|
|
|
6개월 |
3.3% |
23.8% |
1.8% |
12.4% |
2.5% |
20.8% |
|
12개월 |
NA |
NA |
NA |
NA |
4.5% |
23.2% |
a RA 시험 I 24주차, RA 시험 II 26주차, RA 시험 III 24주차 및 52주차
b 40mg 이 약 격주 투여
* p0.01, ACR 20, 50, 70에 대한 모든 시점에서 이 약 vs. 위약
RA 시험 II에서 매주 이 약 40 mg을 투여 받은 환자 또한 6개월차에 통계적으로 유의하게 ACR 20, 50 및 70 반응률에 각각 53.4%, 35.0% 및 18.4% 도달하였다.
RA 시험 III에 대한 ACR 반응 기준의 구성요소 결과는 표 4에 제시되었다. 아래에 제시한 결과는 일반적으로 수행된 각 시험을 대표하는 것이다.
ACR 반응률 및 모든 ACR 반응 기준의 개선은 104주차까지 유지되었다. RA 시험 III에서 2년 동안 이 약 투여군의 24%가 6개월 동안 ACR 70 반응을 유지한 것으로 정의되는 주요 임상 반응에 도달하였다. ACR 반응은 RA 시험 III의 공개 연장 시험에서 이 약 치료를 지속한 환자 중 비슷한 비율의 환자에서 최대 60개월 동안 유지되었다.
RA 시험 III의 공개 연장에서 ACR 반응자 중 대다수의 환자가 최대 10년간의 추적조사 시 반응을 유지했다. 격주 이 약 40 mg 투여군으로 무작위 배정된 환자 207명 중 114명은 5년 동안 격주로 이 약 40 mg을 지속적으로 투여 받았다. 이들 중 86명(75.4%)은 ACR 20 반응, 72명(63.2%)은 ACR 50 반응, 41명(36%)은 ACR 70 반응에 도달하였다. 207명의 환자 중 81명은 10년 동안 이 약 40 mg을 지속적으로 투여 받았다. 이들 중 64명(79.0%)은 ACR 20 반응, 56명(69.1%)은 ACR 50 반응, 43명(53.1%)은 ACR 70 반응에 도달하였다.
표 4: RA 시험 III에서의 ACR 반응 구성요소
|
|
베이스라인 |
24주차 |
52주차 |
베이스라인 |
24주차 |
52주차 |
|
매개변수(중앙값) |
위약/MTX (N=200) |
이 약a/MTX (N=207) | ||||
|
압통 관절 수(0-68) |
26.0 |
15.0 |
15.0 |
24.0 |
8.0* |
6.0* |
|
종창 관절 수(0-66) |
17.0 |
11.0 |
11.0 |
18.0 |
5.0* |
4.0* |
|
의사의 전반적 질병 활성도 평가b |
63.0 |
35.0 |
38.0 |
65.0 |
20.0* |
16.0* |
|
환자의 전반적 질병 활성도 평가b |
53.5 |
39.0 |
43.0 |
52.0 |
20.0* |
18.0* |
|
통증b |
59.5 |
38.0 |
46.0 |
58.0 |
21.0* |
19.0* |
|
장애 척도(HAQ)c |
1.50 |
1.25 |
1.25 |
1.50 |
0.75* |
0.75* |
|
CRP (mg/L) |
10.0 |
9.0 |
9.0 |
10.0 |
4.0* |
4.0* |
|
a 40mg 이 약 격주 투여 b 시각적 아날로그 척도. 0점 = 최고, 100점 = 최악 c 건강 평가 설문조사의 장애 척도. 0점 = 최고, 3점 = 최악. 옷 입기/치장하기, 일어나기, 먹기, 걷기, 뻗기, 잡기, 위생 유지 및 일상 활동 유지 등을 수행할 수 있는 환자의 능력 측정 * p <0.001, 이 약 vs. 위약, 베이스라인으로부터 평균 변화에 기초함 | ||||||
RA 시험 III에서 24주차에 ACR 20 반응에 도달한 환자의 84.7%는 52주차에도 반응을 유지하였다. 다음의 그림은 시험 III 및 II에서이 약에 대한 ACR 20 반응의 지속성을 나타낸다.
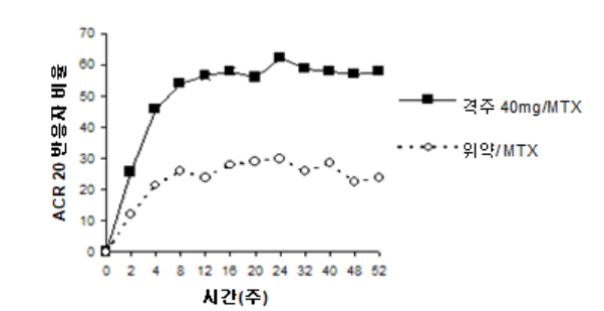
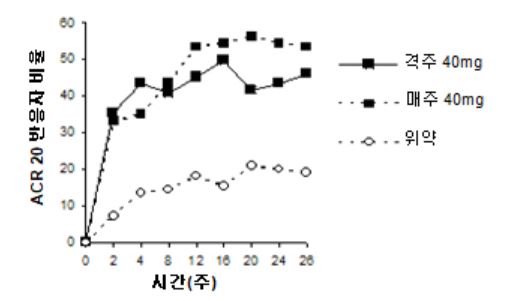
RA 시험 IV
이 약과 함께 표준치료를 투여 받은 환자의 ACR 20 반응은 위약과 함께 표준치료를 투여 받은 환자에 비해 통계적으로 유의하게 우수하였다(p0.001). 이 약과 다른 DMARD의 병용 투여와 관련된 고유의 이상사례는 관찰되지 않았다.
RA 시험 I ‑IV에서 이 약을 투여 받은 환자는 위약을 투여 받은 환자보다 더 빠르고 자주 ACR 20, 50 및 70 반응에 도달하였다. RA 시험 I에서 1주차(첫 시험 방문)의 ACR 20 반응에 대해 이 약을 투여 받은 환자(26.0%)와 위약을 투여 받은 환자(5.0%) 사이에 통계적으로 유의한 차이가 있었다. ACR 20 반응에 대한 통계적으로 유의한 차이는 RA 시험 II, III 및 IV의 2주차(첫 시험 방문)에 이 약을 투여 받은 환자(각각 36.4%, 29.1% 및 33.7%)와 위약을 투여 받은 환자(각각 7.3%, 13.0% 및 8.6%) 사이에서도 확인되었다. 첫 ACR 50 및 70 반응까지의 시간은 4건의 시험 모두에서 유사한 패턴으로 나타났다.
메토트렉세이트를 병용 투여하지 않은 일부 환자에서 매주 40 mg으로 이 약의 투여 빈도를 증가시킴에 따라 추가 이익이 유도될 수 있다. 이는 불완전한 반응을 보인 환자의 투여 빈도를 격주 40 mg에서 매주 40mg으로 증가시킨 장기간 공개 연장 시험에서 확인되었다.
RA 시험 V
메토트렉세이트의 투여 이력이 없는 초기 류마티스 관절염 환자를 대상으로 한 RA 시험 V에서, 이 약과 메토트렉세이트의 병용 치료는 52주차에 메토트렉세이트 단일 치료 및 이 약 단일 치료와 비교하여 더 빠르고 유의하게 큰 ACR 반응을 유발했으며 이 반응은 104주까지 지속되었다(표 5참조).
52주차에 ACR 반응 기준의 모든 개별적인 구성요소는 이 약/메토트렉세이트 치료로 인해 개선되었으며 개선은 104주까지 유지되었다.
2년간의 시험 동안, 이 약/메토트렉세이트 병용 치료를 받은 환자 중 48.5%가 주요 임상 반응(6개월 연속 ACR 70)에 도달했으며, 이에 비해 메토트렉세이트 단일 치료를 받은 환자는 27.2% (p0.001), 이 약 단일 치료를 받은 환자는 24.5% (p0.001)가 주요 임상 반응에 도달하였다.
표 5: RA 시험 V에서의 ACR 반응(환자 비율)
|
반응 |
MTXb N=257 |
이 약c N=274 |
이 약/MTX N=268 |
|
ACR 20 | |||
|
52주차 |
62.6% |
54.4% |
72.8% |
|
104주차 |
56.0% |
49.3% |
69.4% |
|
ACR 50 | |||
|
52주차 |
45.9% |
41.2% |
61.6% |
|
104주차 |
42.8% |
36.9% |
59.0% |
|
ACR 70 | |||
|
52주차 |
27.2% |
25.9% |
45.5% |
|
104주차 |
28.4% |
28.1% |
46.6% |
|
주요 임상 반응a | |||
|
104주차 |
27.2% |
24.5% |
48.5% |
a 주요 임상 반응은 6개월 연속 ACR 70 반응에 도달한 것으로 정의한다.
b p0.05, ACR 20에 대한 이 약/MTX vs. MTX
p0.001, ACR 50, 70 및 주요 임상 반응에 대한 이 약/MTX vs. MTX
c p0.001, 이 약/MTX vs. 이 약
RA 시험 V의 공개 연장시험에서 ACR 반응률은 최대 10년간의 추적조사 시에 유지되었다. 이 약 격주 40 mg 투여군으로 무작위 배정된 환자 542명 중 170명은 10년 동안 이 약 격주 40 mg 투여를 지속하였다. 이들 중 154명(90.6%)은 ACR 20 반응, 127명(74.7%)은 ACR 50 반응, 102명(60.0%)은 ACR 70 반응에 도달하였다. 52주차에 이 약/메토트렉세이트 병용 치료를 받은 환자 중 42.9%가 임상적 관해(질병 활성도 점수[Disease Activity Score; DAS28] ‑CRP 2.6)에 도달하였고, 이에 비해 메토트렉세이트 단일 치료를 받은 환자는 20.6%, 이 약 단일 치료를 받은 환자는 23.4%가 임상적 관해에 도달하였다. 이 약/메토트렉세이트 병용 치료는 최근에 중등증부터 중증의 류마티스 관절염을 진단 받은 환자가 낮은 질병 상태에 도달하는 데 있어 메토트렉세이트(p0.001) 및 이 약 단일 치료(p0.001)보다 통계적 및 임상적으로 우수하였다(표 6참조). 기존에 이 약 단일 치료 또는 이 약/메토트렉세이트 병용 치료로 무작위 배정되어 공개 연장 시험에 등록된 342명의 대상자 중 171명이 10년 동안의 이 약 치료를 완료하였다. 이들 중 109명(63.7%)이 10년차에 질병 관해를 보고하였다.
표 6: RA 시험 V에서의 DAS28 반응
|
DAS28 반응 |
MTX N=257 |
이 약 N=274 |
이 약/MTX N=268 |
|
52주차 | |||
|
베이스라인(평균) |
6.3 |
6.4 |
6.3 |
|
베이스라인으로부터 평균 변화(평균 ± SD) |
-2.8 ± 1.4a |
-2.8 ± 1.5b |
-3.6 ± 1.3 |
|
관해된 환자 비율(DAS28 <2.6) |
20.6%a |
23.4%b |
42.9% |
|
104주차 | |||
|
베이스라인(평균) |
6.3 |
6.3 |
6.3 |
|
베이스라인으로부터 평균 변화(평균 ± SD) |
-3.1 ± 1.4a |
-3.2 ± 1.4b |
-3.8 ± 1.3 |
a p0.001, 이 약/메토트렉세이트 vs. 메토트렉세이트
b p0.001, 이 약/메토트렉세이트 vs. 이 약
방사선학적 반응
RA 시험 III에서 이 약을 투여 받은 환자의 류마티스 관절염 평균 지속 기간은 약 11년이었으며 구조적 관절 손상은 방사선 촬영을 통해 평가하였고 수정된 전체 샤프 점수(modified total Sharp score; TSS) 및 구성요소, 미란(erosion) 점수 및 관절 간격 협착(joint space narrowing score; JSN)의 변화로 결과를 표시하였다. 손/손목 및 발의 앞부분의 방사선 사진은 베이스라인, 6개월 및 12개월에 판독하였다. 12개월 결과는 표 7에 제시한다. 6개월 때 TSS 및 미란 점수의 변화에서 통계적으로 유의한 차이가 관찰되었고 12개월 때 유지되었다. 52주차에 이 약/메토트렉세이트를 투여 받은 환자는 MTX만 단독 투여 받은 환자보다 방사선학적 진행이 더딘 것으로 확인되었다.
표 7: MTX를 병용 투여한 RA 시험 III에서 12개월간의 방사선학적 변화
|
|
위약 N=200 |
이 약a N=207 |
이 약a와 위약간 차이 |
p값 |
|
수정된 전체 샤프 점수의 변화(평균) |
2.7 |
0.1 |
-2.6 |
≤0.001b |
|
미란의 변화(평균) |
1.6 |
0.0 |
-1.6 |
≤0.001 |
|
새로운 미란 없음(환자 비율 %) |
46.2 |
62.9 |
16.7 |
≤0.001 |
|
JSN 점수의 변화(평균) |
1.0 |
0.1 |
-0.9 |
0.002 |
a 격주 40mg 투여
b ranked ANCOVA 분석에 기초함
RA 시험 III의 공개 연장 시험에서 특정 용량의 이 약을 투여 받은 기존의 환자 중 77%는 2년차에 방사선 촬영으로 평가하였다. TSS로 측정한 결과, 환자는 구조적 손상의 억제를 유지했다. 54%의 환자는 구조적 손상의 진행이 없었다(0점 이하의 TSS 변화로 정의). 격주로 이 약 40 mg을 기존에 투여 받은 환자 중 55%는 5년차에 방사선 촬영으로 평가하였다. 환자의 구조적 손상 억제는 지속적이었으며 50%는 구조적 손상의 진행이 없었다(0점 이하의 TSS 변화로 정의).
RA 시험 III의 공개 연장 시험에서 구조적 손상의 진행률 감소는 환자의 하위군에서 8 ‑10년 동안 유지되었다. 격주로 이 약 40 mg을 기존에 투여 받은 환자 207명 중 81명의 환자를 8년차에 방사선 촬영으로 평가하였다. 이들 중 48명의 환자는 구조적 손상의 진행이 없었다(베이스라인으로부터 mTss 변화가 0.5 이하). 격주로 이 약 40 mg을 기존에 투여 받은 환자 207명 중 79명의 환자를 10년차에 방사선 촬영으로 평가하였다. 이들 중 40명의 환자는 구조적 손상의 진행을 보이지 않았다(베이스라인으로부터 mTss 변화가 0.5 이하).
RA 시험 V에서의 구조적 관절 손상은 RA 시험 III과 동일하게 평가하였다. 방사선학적 진행의 억제(TSS, 미란 점수 및 JSN의 변화로 평가)는 52주차와 104주차 모두, 메토트렉세이트 또는 이 약 단일 투여군보다 이 약/메토트렉세이트 병용 투여군에서 높게 관찰되었다(표 8참조).
표 8: RA 시험 V에서 52주차의 방사선학적 평균 변화
|
|
MTXa N=257 (95% 신뢰구간) |
이 약b N=274 (95% 신뢰구간) |
이 약/MTX N=268 (95% 신뢰구간) |
|
전체 샤프 점수 |
5.7 (4.2-7.3) |
3.0 (1.7-4.3) |
1.3 (0.5-2.1) |
|
미란 점수 |
3.7 (2.7-4.7) |
1.7 (1.0-2.4) |
0.8 (0.4-1.2) |
|
JSN 점수 |
2.0 (1.2-2.8) |
1.3 (0.5-2.1) |
0.5 (0-1.0) |
a p0.001, 52주차 및 104주차에서 이 약/MTX vs. MTX
b p0.01, 52주차 이 약/MTX vs. 이 약
p0.001, 104주차 이 약/MTX vs. 이 약
52주 및 104주간의 투여 이후, 질병 진행이 없는(베이스라인으로부터 수정된 전체 샤프 점수의 변화 ≦0.5점) 환자의 비율은 이 약/메토트렉세이트 병용 치료군(각각 63.8% 및 61.2%)이 메토트렉세이트 단일 치료군(각각 37.4%, 33.5%, p0.001)과 이 약 단일 치료군(각각 50.7%, p0.002 및 44.5%, p0.001)에 비해 유의하게 높았다.
RA 시험 V의 공개 연장 시험에서 베이스라인으로부터 10년차까지 수정된 전체 샤프 점수의 평균 변화는 메토트렉세이트 단일 치료군, 이 약 단일 치료군 및 이 약/메토트렉세이트 병용 치료군으로 기존에 무작위 배정된 환자에서 각각 10.8점, 9.2점 및 3.9점이었다. 방사선학적 진행이 없는 환자의 비율은 각각 31.3%, 23.7% 및 36.7%였다.
삶의 질 및 신체적 기능
5건의 적절하고 잘 제어된 시험 모두에서 건강 평가 설문조사(Health Assessment Questionnaire; HAQ)의 장애 척도를 사용하여 건강 관련 삶의 질을 평가하였고, 이는 RA 시험 III에서 사전에 명시된 52주차의 일차 평가변수였다. 4건의 시험 모두에서 이 약의 모든 투여/계획은 베이스라인으로부터 6개월차까지 HAQ의 장애 척도에 대해 위약보다 통계적으로 유의하게 큰 개선을 보였다. RA 시험 III에서 베이스라인으로부터 52주차까지의 HAQ 평균(CI) 개선은 이 약/메토트렉세이트 환자의 경우 ‑0.60 ( ‑0.65, ‑0.55)이었고 위약/메토트렉세이트 환자(p0.001)의 경우 ‑0.25 ( ‑0.33, ‑0.17)였다. 이 약/메토트렉세이트를 투여 받은 환자 중 63%는 시험의 이중눈가림 부분에서 52주차에 0.5 이상의 HAQ 개선에 도달하였다. 이 환자들 중 82%는 104주차까지 개선이 유지되었으며 비슷한 비율의 환자가 공개 연장시험의 260주차(5년)까지 반응이 유지되었다. 신체적 기능이 개선되고 치료를 지속한 대부분의 환자는 공개 연장 시험의 520주차(10년)까지 개선이 유지되었다.
적절하게 잘 제어된 4건의 시험 모두에서 일반적인 건강 관련 삶의 질을 평가하기 위해 약식 건강 조사(SF ‑36) 또한 사용하였다. 4건의 시험 모두에서 이 약의 모든 투여/계획은 베이스라인으로부터 6개월차까지의 SF ‑36 신체적 구성요소 요약 점수에 대해 통계적으로 유의하게 큰 개선을 보였고 이는 RA 시험 III의 52주차에서도 유지되었다. SF ‑36의 평균 개선 또한 156주차(36개월)의 측정 종료 시점까지 유지되었다. 시험 II 및 IV의 SF ‑36 정신적 구성요소 요약 점수 또한 6개월차에 이 약이 위약보다 통계적으로 유의하게 높았다. 이 약을 40mg 격주투여한 4건의 시험 모두에서 베이스라인으로부터 6개월차까지 SF ‑36의 통증 및 활력 영역 점수는 위약과 비교하여 이 약을 투여 받은 환자에서 통계적으로 유의하게 큰 개선을 보였다. 이러한 결과는 분석한 3건의 시험 모두 6개월차에 피로가 통계적으로 유의하게 감소함을 나타낸 만성질환 치료의 기능적 평가(functional assessment of chronic illness therapy; FACIT) 점수로 뒷받침되었으며 RA 시험 III의 52주차에서도 유지되었다.
RA 시험 V에서 HAQ 장애 척도 및 SF ‑36의 신체적 구성요소는 52주차에 메토트렉세이트 단일 치료군 및 이 약 단일 치료군 모두와 비교하여 이 약/메토트렉세이트 병용 치료군에서 더욱 개선되었다(p0.001). 이 개선은 104주차까지 유지되었다. 공개 연장시험을 완료한 250명의 대상자에서 신체적 기능의 개선은 10년의 치료 동안 유지되었다.
투여 부위 통증
통합 교차 RA 시험 VI 및 VII의 경우 아달리무맙 40mg/0.8mL과 아달리무맙 40mg/0.4mL 사이에서 투여 직후 투여 부위 통증에 대해 통계적으로 유의한 차이가 관찰되었다(평균 VAS 3.7cm vs. 1.2cm, 0 ‑10cm 척도, p0.001). 이는 투여 부위 통증의 84% 중앙값 감소를 나타낸다.
건선성 관절염
2건의 무작위배정, 이중눈가림, 위약 대조 시험에서 건선성 관절염 환자 413명을 대상으로 이 약 안전성과 유효성을 평가하였다. PsA 시험 I (M02 ‑518)에는 중등증부터 중증의 활동성 건선성 관절염(>3개의 종창 관절 및 >3개의 압통 관절)이 있는 성인 환자 313명이 등록되었으며 이들은 다음의 유형 중 하나에서 NSAID 치료에 대해 부적절한 반응을 보였다: (1) 원위지절간관절(distal interphalangeal; DIP) 관련(N=23), (2) 다관절형 관절염(류마티스 결절이 없고 건선이 존재)(N=210), (3) 단절성 관절염(N=1), (4) 비대칭성 건선성 관절염(N=77), 또는 (5) 강직성 척추염 유사(N=2). 등록 시 MTX 치료(>1개월 동안 안정적 용량 ≦30 mg/주) 중이었던 환자(313명 중 158명)는 동일 용량으로 메토트렉세이트를 계속 투여 받을 수 있었다. 24주의 이중눈가림 기간 동안 격주로 이 약 40 mg 또는 위약을 투여 받았다. 12주간 수행한 PsA 시험 II (M02 ‑570)는 DMARD 치료에 부적절한 반응을 보인 100명의 환자를 대상으로 하였다. 두 시험의 완료 후 383명의 환자는 공개 연장시험에 등록되어 격주로 40 mg의 이 약을 투여 받았다.
ACR 및 PASI 반응
위약과 비교하여, 이 약의 치료는 질병 활성도 측정에서 개선을 보였다(표 9 및 10 참조). 이 약을 투여 받은 건선성 관절염 환자 중, 첫 방문 시점(2주차)에서 일부 환자의 임상적 반응이 명백하였다. 비록 단절성 관절염 및 강직성 척추염 유사 하위유형에는 극소수의 환자가 등록되었지만 PsA의 각 하위유형 환자에서도 비슷한 반응이 관찰되었다. 베이스라인에서 메토트렉세이트치료를 병용 투여 받거나 병용 투여 받지 않은 환자에서도 반응이 비슷하였다.
최소 3% 체표면적(body surface area; BSA)이 건선과 관련된 환자에서 건선 면적 및 중증도 지수(Psoriatic Area and Severity Index; PASI) 반응을 평가하였다. 24주차에 PASI가 75% 또는 90% 개선에 도달한 환자의 비율은 이 약 투여군(N=69)에서 각각 59%와 42%였고, 이에 비해 위약군(N=69)에서는 각각 1%와 0%였다(p0.001). PASI 반응은 첫 방문 시점(2주차)에 일부 환자에서 명백하였다. 베이스라인에서 메토트렉세이트 치료를 병용 투여 받거나 병용 투여 받지 않은 환자에서도 반응이 비슷하였다.
표 9: 위약대조 건선성 관절염 시험에서의 ACR 반응(환자 비율)
|
반응 |
위약 N=162 |
이 약 N=151 |
위약 N=49 |
이 약 N=51 |
|
|
PsA 시험 I |
PsA 시험 II | ||
|
ACR 20 |
|
|
|
|
|
12주차 |
14% |
58%a |
16% |
39%b |
|
24주차 |
15% |
57%a |
N/A |
N/A |
|
ACR 50 |
|
|
|
|
|
12주차 |
4% |
36%a |
2% |
25%a |
|
24주차 |
6% |
39%a |
N/A |
N/A |
|
ACR 70 |
|
|
|
|
|
12주차 |
1% |
20%a |
0% |
14%b |
|
24주차 |
1% |
23%a |
N/A |
N/A |
a 이 약과 위약 사이의 모든 비교에 대해 p0.001
b 이 약과 위약 사이의 모든 비교에 대해 p0.05
N/A 해당 없음
표 10: 건선성 관절염 PsA 시험 I에서의 질병 활성도 구성요소
|
매개변수: 중앙값 |
베이스라인 |
24주차 |
베이스라인 |
24주차 |
|
|
위약 N=162 |
이 약* N=151 | ||
|
통증이 있는 관절 수a |
23.0 |
17.0 |
20.0 |
5.0 |
|
종창 관절 수b |
11.0 |
9.0 |
11.0 |
3.0 |
|
의사의 전반적 평가c |
53.0 |
49.0 |
55.0 |
16.0 |
|
환자의 전반적 평가c |
49.5 |
49.0 |
48.0 |
20.0 |
|
통증c |
49.0 |
49.0 |
54.0 |
20.0 |
|
장애 척도(HAQ)d |
1.0 |
0.9 |
1.0 |
0.4 |
|
CRPe |
7.8 |
7.4 |
8.0 |
2.1 |
* 중앙값 변화를 기초로 이 약 vs. 위약 비교에 대해 p0.001
a 0 ‑78 척도
b 0 ‑76 척도
c 시각적 아날로그 척도. 0점 = 최고, 100점 = 최악
d 건강 평가 설문조사의 장애 척도. 0점 = 최고, 3점 = 최악. 옷 입기/치장하기, 일어나기, 먹기, 걷기, 뻗기, 잡기, 위생 유지 및 일상 활동 유지 등을 수행할 수 있는 환자의 능력 측정
e 정상 범위: 0 ‑2.87mg/L
최대 136주 동안의 공개 연장시험에서 ACR 반응은 유지되었다.
건선성 관절염 시험에서 방사선학적 변화를 평가하였다. 손, 손목 및 발의 방사선 사진은 환자가 이 약 또는 위약을 투여 받은 이중눈가림 기간의 베이스라인과 24주차, 그리고 모든 환자가 공개 연장 시험의 이 약을 투여 받은 48주차에 수집하였다. 원위지절간관절을 포함한 수정된 전체 샤프 점수(modified Total Sharp Score; mTss)(즉, 류마티스 관절염에서 사용한 TSS와 동일하지 않음)를 사용하였다.
이 약 치료는 위약 치료와 비교하여 말초 관절 손상의 진행률을 감소시켰고, 이는 베이스라인으로부터 mTss 점수(평균 ± SD) 변화로 측정하였으며 위약군의 경우 0.8 ± 2.5 (24주차), 이 약 투여군의 경우 0.0 ± 1.9 (48주차)였다(p0.001).
베이스라인으로부터 48주차까지 방사선학적 진행이 없는 이 약 투여군(n=102) 중, 84%는 144주간의 치료 동안 방사선학적 진행을 보이지 않았다.
삶의 질 및 신체적 기능
이 약을 투여 받은 환자는 24주차에 위약과 비교하여 HAQ 및 약식 건강 조사(SF ‑36) 평가 결과 신체적 기능에 있어서 통계적으로 유의한 개선이 확인되었다. 개선된 신체적 기능은 최대 136주차의 공개 연장 시험동안 지속되었다.
축성 척추관절염 임상시험
강직성 척추염
활동성 강직성 척추염(ankylosing spondylitis; AS)이 있는 성인 환자 393명을 대상으로 2건의 무작위배정, 24주 이중눈가림, 위약대조 시험에서 이 약 40 mg의 격주 투여에 대한 안전성과 유효성을 평가하였다. 2건 중 규모가 큰 시험(AS 시험 I 또는 M03 ‑607)에는 활동성 강직성 척추염이 있고 기존의 치료에 부적절한 반응을 보인 성인 환자 315명이 등록되었다. 활동성 강직성 척추염은 다음의 3가지 기준 중 최소 2가지를 충족하는 것으로 정의한다: (1) 강직성 척추염 질병 활성도 지수(Bath AS disease activity index; BASDAI] 점수 ≧4 cm, (2) 전체 요통에 대한 시각적 아날로그 점수(visual analog score; VAS) ≧40 mm, (3) 조조 강직 ≧1시간. 눈가림 기간 후 공개 연장시험 기간 동안 환자는 최대 236주의 추가 기간 동안 이 약 40mg을 격주로 피하투여 받았다.
그 결과 이 약을 투여 받은 환자는 위약을 투여 받은 환자와 비교하여 강직성 척추염의 징후 및 증상이 통계적으로 유의하게 개선되었다. 그림 4와 표 11 및 12에 제시한 바와 같이, 질병 활성도 측정으로 확인한 유의한 개선은 2주차에 처음 관찰되었고 24주까지 유지되었다.
전체 척추 강직증이 있는 환자는 이 시험에 포함되었다(n=11). 이 환자들의 반응은 전체 강직증이 없는 환자들과 유사하였다.
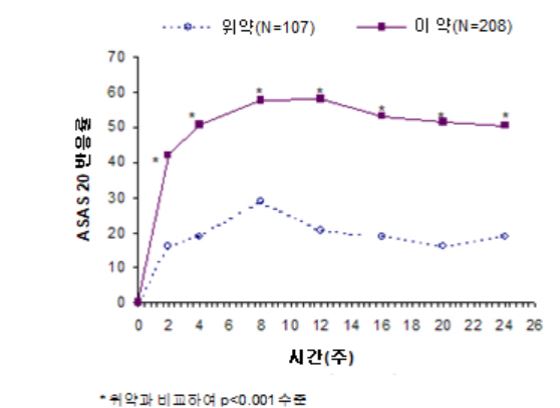
표 11: 위약대조 AS 시험에서의 유효성 결과 ‑ AS 시험 I 징후 및 증상 감소
|
반응 |
위약 N=107 |
이 약 N=208 |
|
ASASa 20 |
|
|
|
2주차 |
16% |
42%*** |
|
12주차 |
21% |
58%*** |
|
24주차 |
19% |
51%*** |
|
ASAS 50 |
|
|
|
2주차 |
3% |
16%*** |
|
12주차 |
10% |
38%*** |
|
24주차 |
11% |
35%*** |
|
ASAS 70 |
|
|
|
2주차 |
0% |
7%** |
|
12주차 |
5% |
23%*** |
|
24주차 |
8% |
24%*** |
|
BSADAIb 50 |
|
|
|
2주차 |
4% |
20%*** |
|
12주차 |
16% |
45%*** |
|
24주차 |
15% |
42%*** |
***,** 2, 12, 24주차에서 이 약과 위약 사이의 모든 비교에 대해 p0.001, p0.01에서 통계적으로 유의함
a 강직성 척추염에서의 평가
b 강직성 척추염 활동 지수
위약을 투여 받은 환자 6%와 비교하여 이 약을 투여 받은 환자 22%가 24주차에 낮은 수준의 질병 활성도(4개의 ASAS 반응 매개변수 각각의 수치가 20 [0 ‑100mm 척도])에 도달하였다 (p0.001).
표 12: 강직성 척추염 질병 활성도의 구성요소
|
|
베이스라인 평균 |
24주차 평균 |
베이스라인 평균 |
24주차 평균 |
|
|
위약 N=107 |
이 약 N=208 | ||
|
ASAS 20 반응 기준* |
|
|
|
|
|
환자의 전반적 질병 활성도 평가a* |
65 |
60 |
63 |
38 |
|
전체 요통* |
67 |
58 |
65 |
37 |
|
염증b* |
6.7 |
5.6 |
6.7 |
3.6 |
|
BASFIc* |
56 |
51 |
52 |
34 |
|
BASDAId 점수* |
6.3 |
5.5 |
6.3 |
3.7 |
|
BASMIe 점수* |
4.2 |
4.1 |
3.8 |
3.3 |
|
이주부터 벽까지의 거리(cm) |
15.9 |
15.8 |
15.8 |
15.4 |
|
요추 굴곡(cm) |
4.1 |
4.0 |
4.2 |
4.4 |
|
목 회전(각도) |
42.2 |
42.1 |
48.4 |
51.6 |
|
요추방향 굴곡(cm) |
8.9 |
9.0 |
9.7 |
11.7 |
|
복사뼈 사이의 거리(cm) |
92.9 |
94.0 |
93.5 |
100.8 |
|
CRPf* |
2.2 |
2.0 |
1.8 |
0.6 |
a 시각적 아날로그 척도(VAS) 0점 = “없음” 및 100점 = “중증”으로 측정 시에, 최소 20% 및 10점이 개선된 대상자의 비율
b BASDAI(‘d’에서 정의) 5번 및 6번 질문의 평균
c 강직성 척추염 기능 지수
d 강직성 척추염 질병 활성도 지수
e 강직성 척추염 운동능력 지수
f C ‑반응성 단백질(mg/dL)
* 24주차에 이 약과 위약 사이의 모든 비교에 대해 통계적으로 유의함
2건의 시험 중 규모가 작은 무작위, 이중눈가림, 위약대조 시험(AS 시험 II, M03 ‑606)의 강직성 척추염 환자 82명에서도 비슷한 경향이 확인되었다.
환자 보고 결과는 2건의 강직성 척추염 시험 모두에서 일반적인 건강 상태 설문조사 SF ‑36 및 질병 특이적 강직성 척추염 삶의 질 설문조사(Ankylosing Spondylitis Quality of Life Questionnaire; ASQoL)를 사용하여 평가하였다. 12주차의 SF ‑36 신체적 구성요소 점수는 이 약을 투여 받은 환자(평균 변화 6.93)가 위약을 투여 받은 환자(평균 변화 1.55, p0.001)에 비해 유의하게 개선되었으며 이는 24주차까지 유지되었다(평균 변화 7.44 vs. 1.85).
ASQoL의 결과는 전반적인 삶의 질 개선을 입증하는 이 결과를 뒷받침한다. 이 약을 투여 받은 환자(평균 변화 ‑3.15, p0.001)는 위약을 투여 받은 환자(평균 변화 ‑0.95, p0.001)와 비교하여 12주차에 통계적으로 유의하게 개선되었으며, 이는 24주차까지 유지되었다(평균 변화 ‑3.58 vs ‑1.06).
비방사선학적 축성 척추관절염(방사선학적으로 강직성 척추염이 확인되지 않는 축성 척추관절염)
비방사선학적 축성 척추관절염(non ‑radiographic axial spondyloarthritis; nr ‑asSpA) 환자를 대상으로 2건의 무작위, 이중눈가림, 위약대조 시험에서 이 약의 안전성 및 유효성을 평가하였다. nr ‑axSpA I 시험에서는 활동성 nr ‑axSpA가 있는 환자를 평가하였다. nr ‑axSpA II 시험은 공개 연장 이 약 치료 동안 질병 관해에 도달한 활동성 nr ‑axSpA 환자를 대상으로 한 치료 중단 시험이었다.
nr ‑axSpA I 시험
무작위배정, 12주 이중눈가림, 위약대조 시험인 nr ‑axSpA I 시험에서는 활동성 nr ‑axSpA(질병 활성도의 평균 베이스라인 점수[강직성 척추염 질병 활성도 지수(BASDAI)]가 이 약 투여군의 경우 6.4, 위약군의 경우 6.5) 환자 185명을 대상으로 이 약 40 mg 격주 투여를 평가하였으며, 이 환자들은 ≧1개의 NSAID에 대해 부적절한 반응 또는 불내성이 있거나 NSAID가 금지된 환자였다. 여기에 포함된 환자들은 ASAS 축성 SpA 기준에 따라 분류되었고, 강직성 척추염에 대해 수정된 뉴욕 기준을 충족하는 환자 및 건선이나 건선성 관절염이 있는 환자는 제외하였다. 일차 유효성 평가변수는 12주차에 ASAS 40 반응에 도달한 환자 비율이었다.
베이스라인에서 33명(18%)의 환자는 질병 조절 항류마티스제를 병용 투여 받았고 146명(79%)은 NSAID를 병용 투여 받았다. 이중눈가림 기간 후 공개 연장 기간 동안 환자는 최대 144주간 추가적으로 이 약 40 mg을 격주 피하투여 받았다. 12주차 결과에 따르면 전체 치료군 및 양성 MRI 또는 증가된 CRP를 보인 환자 모두에서 위약과 비교하여 이 약을 투여 받은 환자의 활동성 nr ‑axSpA 징후 및 증상이 통계적으로 유의하게 개선되었다(표 13). nr ‑axSpA의 징후 및 증상이 감소했음을 입증한 변수는 24주차 및 68주차에서도 지속되거나 계속 개선되었으며 156주차까지 유지되었다.
표 13: 위약대조 시험 nr ‑axSpA I에서의 유효성 결과#
|
이중눈가림 12주차에서의 반응 |
위약 N=94 |
이 약 N=91 |
|
ASASa 40 |
15% |
36%*** |
|
ASAS 20 |
31% |
52%** |
|
ASAS 5/6 |
6% |
31%*** |
|
ASAS 부분 관해 |
5% |
16%* |
|
BASDAIb 50 |
15% |
35%** |
|
ASDASc,d,e |
-0.3 |
-1.0*** |
|
ASDAS 비활성 질병 |
4% |
24%*** |
|
SF-36 PCSd,f |
2.0m |
5.5** |
|
HAQ-Sd,g |
-0.1 |
-0.3* |
|
hs-CRPd,h,i |
-0.3 |
-4.7*** |
|
SPARCCj MRI 천장 관절d,k |
-0.6 |
-3.2** |
|
SPARCC MRI 척추d,l |
-0.2 |
-1.8** |
|
a 국제 척추관절염 학회의 평가 b 강직성 척추염 질병 활성도 지수 c 강직성 척추염 질병 활성도 점수 d 베이스라인으로부터의 평균 변화 e 위약 n=91, 이 약 n=87 f 약식-36 건강 상태 설문조사TM 버전 2 신체적 구성요소 요약 점수 g 척추관절증에 대해 수정한 건강 평가 설문조사 h 고감도 C-반응 단백질(mg/L) i 위약 n=73, 이 약 n=70 j 캐나다의 척추관절염 연구 컨소시엄 k 위약 및 이 약 n=84 l 위약 n=82, 이 약 n=85 m n=93 *** p값 < 0.001 ** p값 < 0.01 * p값 < 0.05 # HAQ-S와 hs-CRP의 경우 이전 관찰치 적용(Last observation carried forward; LOCF) 분석, SF-36과 SPARCC MRI 점수의 경우 관찰된 사례 분석, 기타 모든 범주형 변수의 경우 비응답자 결측값 대체(non-responder imputation; NRI) 분석 | ||
염증의 억제
이 약을 투여 받은 환자에서 hs ‑CRP와, 천장관절 및 척추의 MRI로 측정한 염증 징후의 유의한 개선은 각각 156주차와 104주차까지 유지되었다. 천장관절에 대한 SPARCC MRI는 131명의 환자에서 이용 가능했고 척추에 대한 SPARCC MRI는 130명의 환자에서 이용 가능했으며 베이스라인으로부터 104주차까지의 평균 변화는 각각 ‑3.8 및 ‑1.4였다.
삶의 질 및 신체적 기능
건강 관련 삶의 질 및 신체적 기능은 HAQ ‑S 및 SF ‑36 설문조사를 사용하여 평가하였다. 이 약은 위약과 비교하여 베이스라인으로부터 12주차까지 HAQ ‑S 전체 점수 및 SF ‑36 신체적 구성요소 요약(Physical Component Summary; PCS)에 대해 통계적으로 유의하게 큰 개선을 보였다. SF ‑36 PCS 점수 및 HAQ ‑S 전체 점수 결과는 각각 52주차, 68주차 및 156주차까지 지속되었다.
nr ‑axSpA II 시험
≧2개의 NSAID에 대해 부적절한 반응을 보이거나 NSAID에 불내성 또는 금지 사유가 있는 활동성 nr ‑axSpA(평균 베이스라인 질병 활성도[BASDAI]가 7.0) 환자 673명은 nr ‑axSpA II 공개 연장 시험의 표지 기간에 등록되어 28주간 격주로 이 약 40mg을 투여 받았다. 이 환자들은 천장관절이나 척추의 염증에 대해 MRI 또는 증가된 hs ‑CRP와 같은 객관적 증거를 가지고 있었다. 공개 연장 시험 기간 동안 최소 12주차에 지속 관해에 도달한 환자(N=305)(16, 20, 24 및 28주차 ASDAS 1.3)는 이중눈가림, 위약대조 기간(총 시험 기간 68주)의 추가 40주 동안 이 약 40mg 격주 투여군(N=152) 또는 위약군(N=153)으로 무작위 배정되었다. 이중눈가림 기간 동안 flared대상자는 최소 12주 동안의 이 약 40mg 격주 구조요법이 허용되었다.
일차 유효성 평가변수는 시험의 68주차까지 재발이 없는 환자 비율이었다. 재발은 4주 간격의 2회 연속 방문에서 ASDAS ≧ 2.1로 정의하였다. 이 약을 투여 받은 대다수의 환자는 위약을 투여 받은 환자와 비교하여 이중눈가림 기간 동안 질병 재발이 없었다(70.4% vs. 47.1%, p0.001)(그림 5).

그림 5: nr ‑axSpA II에서 재발까지의 시간을 요약한 카플란 ‑메이어 곡선
치료 중단에 배정된 그룹에서 재발한 환자 68명 중 65명은 12주간의 이 약 구조요법을 완료하였고 이 중 37명(56.9%)은 공개 연장 시험을 재개하고 12주 후에 다시 질병 관해에 도달하였다(ASDAS 1.3).
68주까지 지속적으로 이 약을 투여 받은 환자는 이중눈가림 기간 동안 치료 중단으로 배정된 환자와 비교하여 활동성 nr ‑axSpA의 징후 및 증상에 대해 통계적으로 유의하게 큰 개선을 보였다(표 14).
표 14: nr ‑axSpA II 위약대조 기간에서의 유효성 결과
|
이중눈가림 68주차에서의 반응 |
위약 N=153 |
이 약 N=152 |
|
ASASa,b 20 |
47.1% |
70.4%*** |
|
ASASa,b 40 |
45.8% |
65.8%*** |
|
ASASa 부분 관해 |
26.8% |
42.1%** |
|
ASDASc 비활성 질병 |
33.3% |
57.2%*** |
|
부분 재발d |
64.1% |
40.8%*** |
|
a 국제 척추관절염 학회의 평가 b 환자가 활동성 질병을 가지고 있는 경우 베이스라인은 공개라벨 베이스라인으로 정의한다. c 강직성 척추염 질병 활성도 점수 d 부분 재발은 연속 2회 방문에서 ASDAS가 ≥1.3이고 <2.1인 것으로 정의한다. *** p값 < 0.001 ** p값 < 0.01 | ||
크론병
무작위배정, 이중눈가림, 위약대조 시험에서 중등증부터 중증의 활동성 크론병(크론병 활성도 지수[Crohn’s Disease Activity Index; CDAI]가 ≧220 및 ≦450)이 있는 성인 환자를 대상으로 이 약 다중 투여의 안전성과 유효성을 평가하였다. 안정적 용량의 아미노살리실산염, 코르티코스테로이드 및/또는 면역조절제의 투여가 허용되었으며 79%의 환자는 이러한 약물 중 최소 하나의 투여를 지속하였다.
2건의 시험에서 임상적 관해(CDAI 150의 정의)의 유도를 평가하였다. CD 시험 I (M02 ‑403)에서 TNF 차단제 투여 이력이 없는 환자 299명은 다음의 치료군 중 하나로 무작위 배정되었다: 0주차와 2주차에 위약을 투여 받는 위약군, 0주차에 이 약 160 mg과 2주차에 이 약 80 mg을 투여 받는 160/80 투여군, 0주차에 이 약 80 mg과 2주차에 이 약 40 mg을 투여 받는 80/40 투여군, 0주차에 이 약 40mg과 2주차에 이 약20 mg을 투여 받는 40/20 투여군. 임상 결과는 4주차에 평가하였다.
두 번째 유도 시험인 CD 시험 II (M04 ‑691)에서, 인플릭시맙 치료에 반응을 보이지 않거나 불내성이 있는 325명의 환자는 0주차에 이 약 160 mg과 2주차에 이 약 80 mg을 투여 받거나 0주차와 2주차에 위약을 투여 받는 집단으로 무작위 배정되었다. 임상 결과는 4주차에 평가하였다.
CD 시험 III (M02 ‑404)에서 임상적 관해의 유지를 평가하였다. 이 시험에서 활동성 질병이 있는 환자 854명은 공개라벨 이 약을 0주차에 80 mg, 2주차에 40 mg 투여 받았다. 그런 다음 환자는 4주차에 격주 40 mg 이 약 투여군, 매주 40 mg 이 약 투여군, 또는 위약군으로 무작위 배정되었다. 전체 시험 기간은 56주였다. 4주차에 임상 반응을 보인 환자(CR ‑70 = CDAI ≧70 감소)는 4주차에 임상 반응을 보이지 않은 환자와 별도로 계층화하여 분석하였다.
임상적 관해의 유도
TNF 차단제 투여 이력이 없거나(CD 시험 I), 또는 인플릭시맙 치료에 반응을 보이지 않거나 불내성이 있는 것(CD 시험 II)과는 상관 없이 160/80 mg 이 약을 투여 받은 환자의 대부분은 4주차에 위약과 비교하여 임상적 관해를 유도하였다(표 15 참조).
표 15: CD 시험 I 및 CD 시험 II에서 임상적 관해의 유도(환자 비율)
|
|
CD 시험 I |
CD 시험 II | ||
|
|
위약 N=74 |
이 약 160/80mg N=76 |
위약 N=166 |
이 약 160/80mg N=159 |
|
4주차 |
|
|
|
|
|
임상적 관해 |
12% |
36%* |
7% |
21%* |
|
임상 반응 |
34% |
58%** |
34% |
52%** |
|
임상적 관해는 CDAI 점수 <150으로 정의한다. 임상적 반응은 최소 70점의 CDAI 감소로 정의한다. * 비율에 대한 이 약 vs. 위약 쌍비교, p <0.001 ** 비율에 대한 이 약 vs. 위약 쌍비교, p <0.01 | ||||
임상적 관해의 유지
CD 시험 III의 4주차에 환자의 58%(499명/854명)가 임상 반응을 보였고 일차 분석에서 평가되었다. 4주차에서 임상 반응을 보인 환자의 대부분이 26주차와 56주차의 위약 유지군 환자와 비교하여 이 약 40 mg 격주 유지군에서 임상적 관해에 도달하였다(표 16 참조). 매주 이 약 치료를 받은 집단은 이 약을 격주로 투여 받은 집단과 비교하여 유의하게 높은 관해율을 입증하지 못했다.
표 16: CD 시험 III에서 임상적 관해의 유지(환자 비율)
|
평가변수 |
위약 N=170 |
격주 40mg 이 약 N=172 |
매주 40mg 이 약 N=157 |
|
26주차 | |||
|
임상적 관해 |
17% |
40%* |
47%* |
|
임상 반응 |
28% |
54%* |
56%* |
|
56주차 | |||
|
임상적 관해 |
12% |
36%* |
41%* |
|
임상 반응 |
18% |
43%* |
49%* |
임상적 관해는 CDAI 점수 150으로 정의한다. 임상적 반응은 최소 70점의 CDAI 감소로 정의한다.
* 비율에 대한 이 약 vs. 위약 쌍비교, p0.001
시험 동안 관해에 도달한 4주차 반응을 보인 환자 중 격주로 이 약을 투여 받은 환자가 위약 유지군의 환자보다 더 장기간 관해를 유지하였다. 56주차에 질병과 관련된 입원 및 수술은 위약과 비교하여 이 약을 투여 받은 환자에서 통계적으로 유의하게 감소하였다.
CD 시험 I의 환자 117명/276명과 CD 시험 II 및 III의 환자 272명/777명은 최소 3년의 공개라벨 이 약 치료 동안 추적되었다. 각각 88명(75.2%)과 189명(69.5%)의 환자는 임상적 관해가 지속되었다. 임상 반응(CR ‑70)은 각각 107명(91.5%)과 248명(91.2%)의 환자에서 유지되었다.
117명/854명(CD 시험 III)은 스크리닝과 베이스라인 시점 모두에서 배출 누공(draining fistula)을 가지고 있었다. 누공 치유의 평가를 위해 시험에서 사용한 아달리무맙의 두 투여량 모두에 대한 데이터를 통합하였다. 26주차에 누공 치유된 환자의 비율은 아달리무맙을 투여 받은 환자(21명/70명[30.0%])가 위약을 투여 받은 환자(6명/47명[12.8%])에 비해 통계적으로 유의하게 높았다. 완전한 누공 치유는 아달리무맙 투여군과 위약 투여군의 각각 23명/70명(32.9%)과 6명/47명(12.8%)에서 56주차까지 유지되었다.
135명의 환자가 등록된 내시경 시험(M05 ‑769)에서 점막 치유에 대한 이 약의 영향을 확인하였다. 12주차의 점막 치유는 이 약 투여군 중 27.4%, 위약 투여군 중 13.1%에서 확인되었고(p=0.056) 52주차의 점막 치유는 이 약을 투여 받은 환자 중 24.2%, 위약을 투여 받은 환자 중 0%에서 확인되었다(p0.001).
삶의 질
CD 시험 I과 CD 시험 II에서 이 약 80/40 mg 및 160/80 mg 투여군으로 무작위 배정된 환자는 위약군 환자에 비해 질병 특이적 염증성 장질환 설문조사(inflammatory bowel disease questionnaire; IBDQ) 전체 점수에서 통계적으로 유의한 개선을 보였으며, CD 시험 III의 경우 26주차와 56주차에 위약군 환자와 비교하여 아달리무맙 치료군 사이에서도 이러한 개선이 관찰되었다.
궤양성 대장염
2건의 무작위, 이중눈가림, 위약대조 시험에서 중등증부터 중증의 활동성 궤양성 대장염(Mayo 점수 6 ‑12점, 내시경 항목 점수 2 ‑3점)이 있는 성인 환자를 대상으로 이 약 다중 투여의 안전성과 유효성을 평가하였다. 안정적 용량의 아미노살리실산염, 코르티코스테로이드 및/또는 면역조절제의 병용 투여가 허용되었다.
UC ‑I 시험에서 임상적 관해의 유도(Mayo ≦2점, >1점인 항목별 점수는 없는 것으로 정의)를 평가하였다. UC ‑I 시험에서 TNF 길항제 투여 이력이 없는 환자 390명은 0주차와 2주차에 위약 투여군, 0주차에 160 mg 이 약투여 후 2주차에 80 mg 투여군, 또는 0주차에 80 mg 이 약 투여 후 2주차에 40 mg 투여군으로 무작위 배정되었다. 2주차 후에 두 아달리무맙 투여군의 환자는 아달리무맙을 격주로 40 mg 투여 받았다. 임상적 관해는 8주차에 평가되었다.
UC ‑II 시험에서 248명의 환자는 0주차에 160 mg 이 약, 2주차에 80 mg 및 이후 격주로 40 mg 투여 받았으며 246명의 환자는 위약을 투여 받았다. 임상 결과는 8주차에 관해의 유도, 52주차에 관해의 유지에 대해 평가하였다.
160/80 mg 이 약을 투여 받은 대상자는 위약과 비교하여 UC ‑I 시험(각각 18% vs. 9%, p=0.031)과 UC ‑II 시험(각각 17% vs. 9%, p=0.019)에서 통계적으로 유의하게 큰 비율로 8주차에 임상적 관해에 도달하였다. UC ‑II 시험의 8주차에 관해에 도달했던 이 약 투여군 중 21명/41명(51%)은 52주차에 관해를 유지하였다.
전체 UC ‑II 시험군과 전체 Mayo 점수당 치료 8주차에 반응한 환자의 결과는 표 17에 제시한다.
표 17: UC ‑II 시험에서 반응, 관해 및 점막 치유(환자 비율)
|
|
위약
|
격주 40 mg 이 약
|
이 약 160/80/40 mg 8주차 반응자 |
|
|
N=246 |
N=248 |
N=125 |
|
52주차 | |||
|
임상 반응 |
18% |
30%* |
47% |
|
임상적 관해 |
9% |
17%* |
29% |
|
점막 치유 |
15% |
25%* |
41% |
|
≥90일 동안 스테로이드 사용 없는 관해율a |
6% (N=140) |
13%* (N=150) |
20% (N=90) |
|
8주차 및 52주차 | |||
|
지속적 반응 |
12% |
24%** |
- |
|
지속적 관해 |
4% |
8%* |
- |
|
지속적 점막 치유 |
11% |
19%* |
- |
|
임상적 관해는 Mayo 점수가 ≤2이고 >1인 항목별 점수가 없는 것으로 정의한다. * 비율에 대한 이 약 vs. 위약 쌍비교, p <0.05 ** 비율에 대한 이 약 vs. 위약 쌍비교, p <0.001 a 베이스라인에서 코르티코스테로이드를 투여 받은 환자 중 | |||
UC I와 UC II 시험의 통합 분석에서 입원의 모든 원인 및 UC와 관련된 입원율이 통계적으로 유의하게 감소했음을 관찰하였다.
UC ‑II 시험에서 약 40%의 환자가 인플릭시맙을 통한 항 ‑TNF 치료에 실패한 이력이 있었다. 이러한 환자에서 아달리무맙의 효능은 항 ‑TNF 투여 이력이 없는 환자와 비교하여 감소하였다. 항 ‑TNF 치료에 실패 이력이 있는 환자 중 52주차 관해율은 위약군에서 3%, 아달리무맙군에서 10%에 도달하였다.
UC 시험 I 및 II의 환자는 공개라벨 장기간 확장 시험(UC ‑III)으로 전환할 수 있는 선택권이 있었다. 3년간의 이 약 치료 후 74%(268명/360명)의 환자에서 부분 Mayo 점수당 임상적 관해가 지속되었고 최소 4년간 이 약 치료를 받은 환자의 경우 75%(97명/130명)가 부분 Mayo 점수당 임상적 관해가 지속되었다. 1년 이상의 치료 후 반응 도달에 실패한 환자는 매주 40 mg으로 투여 빈도를 증가시킴으로써 치료 이익을 받을 수 있었다.
삶의 질
UC 시험 II에서 이 약 160/80 mg 투여군으로 무작위 배정된 환자는 위약과 비교하여 52주차에 질병 특이적 염증성 장질환 설문조사(IBDQ) 전체 점수의 개선에 도달하였다(p=0.007).
베체트 장염
기존의 치료(스테로이드 또는 면역억제제)에 부적절한 반응을 보인 장내 베체트병이 있는 일본인 환자 대상의 공개, 비통제(uncontrolled) 시험에서 24주차에 현저한 개선율은 45.0%(9명/20명)였다. 현저한 개선은, 종합적인 질병 평가 척도를 사용하여 위장 증상의 전반적 평가와 내시경 평가 점수 모두 ≦1점인 환자의 비율로 정의하였고 이는 궤양의 크기 및 위장 증상 개선을 둘 다 반영한다.
판상 건선
무작위배정, 이중눈가림 시험에서 전신 치료 또는 광선치료의 대상자인 만성 판상 건선( ≧10% BSA 관련, 건선 영역 및 중증도 지수[Psoriasis Area and Severity Index; PASI] ≧12 또는 ≧10)이 있는 성인 환자에서 이 약의 안전성과 유효성을 시험하였다. 건선 시험 I과 II에 등록된 환자 중 73%는 전신 치료나 광선치료를 받은 이력이 있었다. 또한 무작위 배정된 이중눈가림 시험(건선 시험 III)에서 전신 치료의 대상자인 손 및/또는 발의 건선을 동반한 중등증부터 중증의 만성 판상형 건선이 있는 성인 환자에서도 이 약의 안전성과 유효성을 시험하였다.
건선 시험 I (M03 ‑656)은 3개의 치료 기간 동안 1,212명의 환자를 평가하였다. A 기간에 환자는 위약을 투여 받거나 초기 용량 80 mg의 이 약을 투여 받은 후 일주일 뒤부터 격주로 40 mg을 투여 받았다. 16주간의 치료 후 최소 PASI 75 반응(베이스라인과 비교하여 최소 75%의 PASI 점수 개선)에 도달한 환자는 B 기간에 진입하여 공개라벨 40 mg 이 약을 격주로 투여 받았다. 33주차에 ≧PASI 75 반응이 유지되고 기존에 A 기간에서 활성 치료로 무작위 배정된 환자는 C 기간에 무작위 재배정되어 추가 19주 동안 격주로 40 mg 이 약 또는 위약을 투여 받았다. 모든 치료군에서 평균 베이스라인 PASI 점수는 18.9점이었고 베이스라인에서 의사의 전반적 평가(Physician’s Global Assessment; PGA) 점수의 범위는 “중등증”(환자의 53%) ‑“중증”(41%) ‑“매우 중증”(6%)이었다.
건선 시험 II (M04 ‑716)에서는 271명의 환자에서 메토트렉세이트 및 위약에 대해 이 약의 안전성과 유효성을 비교하였다. 환자는 위약, 메토트렉세이트 초기 용량인 7.5mg을 투여 받은 후 12주차까지 최대 25 mg으로 증량하여 투여 받거나, 이 약 초기 용량 80 mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 40 mg을 투여 받았다. 16주 이상의 치료를 통해 이 약과 메토트렉세이트를 비교한 이용가능한 데이터는 없다. MTX를 투여 받고 8주차 및/또는 12주차에 ≧PASI 50 반응에 도달한 환자는 추가로 투여량을 증량하지 않았다. 모든 치료군에서 평균 베이스라인 PASI 점수는 19.7점이었고 베이스라인 PGA 점수의 범위는 “경증”(1%) ‑“중등증”(48%) ‑“중증”(46%) ‑“매우 중증”(6%)이었다.
임상2상 및 임상3상 건선 시험에 모두 참여한 환자는 이 약을 추가로 최소 108주 동안 투여 받는 공개라벨 확장 시험(M03 ‑658)에 등록할 수 있었다.
건선 시험 I과 II에서 일차 평가변수는 베이스라인으로부터 16주차에 PASI 75 반응에 도달한 환자의 비율이었다(표 18 및 19 참조).
표 18: 16주차의 Ps 시험 I (REVEAL) 유효성 결과
|
|
위약 N=398 n(%) |
격주 이 약 40mg N=814 n(%) |
|
≥PASI 75a |
26 (6.5) |
578 (70.9)b |
|
PASI 100 |
3 (0.8) |
163 (20.0)b |
|
PGA: 개선/최소 |
17 (4.3) |
506 (62.2)b |
|
a PASI 75 반응에 도달한 환자 비율은 중심이 보정된 비율로 계산하였다. b p <0.001, 이 약 vs. 위약 | ||
표 19: 16주차의 Ps 시험 II (CHAMPION) 유효성 결과
|
|
위약 N=53 n(%) |
MTX N=110 n(%) |
격주 이 약 40mg N=108 n(%) |
|
≥PASI 75 |
10 (18.9) |
39 (35.5) |
86 (79.6)a,b |
|
PASI 100 |
1 (1.9) |
8 (7.3) |
18 (16.7)c,d |
|
PGA: 개선/최소 |
6 (11.3) |
33 (30.0) |
79 (73.1)a,b |
|
a p <0.001, 이 약 vs. 위약 b p <0.001, 이 약 vs. 메토트렉세이트 c p <0.01, 이 약 vs. 위약 d p <0.05, 이 약 vs. 메토트렉세이트 | |||
건선 시험 I에서 PASI 75 반응에 도달하여 33주차에 위약으로 무작위 재배정된 환자의 28%는 이 약을 지속적으로 투여 받은 환자의 5%와 비교하여(p0.001) “적절한 반응의 실패(loss)”을 경험하였다(베이스라인과 비교하여 PASI 50 반응을 나타내고 33주차와 비교하여 PASI 점수가 6점 이상 감소한, 33주 후 52주차 이하의 PASI 점수). 위약으로 무작위 재배정된 후 적절한 반응 도달에 실패하여 공개라벨 확장 시험에 등록된 환자 중 38% (25명/66명)과 55% (36명/66명)는 각각 12주와 24주의 재치료를 받은 후 PASI 75 반응에 다시 도달하였다.
16주차와 33주차의 PASI 75 반응자 총 233명은 건선 시험 I에서 52주 동안 지속적으로 이 약을 투여 받았으며 공개라벨 확장 시험에서 이 약을 지속적으로 투여 받았다. 이 환자들에서 PASI 75와 PGA 개선 또는 최소 반응률은 공개라벨에서의 추가 108주 치료 후 각각 74.7%와 59.0%였다(총 160주).
건선 시험 II에서 이 약 치료로 무작위 배정된 총 94명은 공개라벨 확장 시험에서 이 약을 지속적으로 투여 받았다. 이 환자들에서 PASI 75와 PGA 개선 또는 최소 반응률은 공개라벨에서의 추가 108주 치료 후 각각 58.1%와 46.2%였다(총 124주).
총 347명의 안정적인 반응자가 공개라벨 확장 시험에서 중단 및 재치료 평가에 참여하였다. 재발까지의 시간 중앙값(PGA가 “중등증” 또는 최악으로 감소)은 약 5개월이었다. 중단 기간 동안 반동현상(rebound)을 경험한 환자는 없었다. 재치료 기간에 등록된 환자 중 76.5% (218명/285명)은 중단 동안의 재발 여부와 관계 없이 재치료 16주 후 PGA “개선” 또는 “최소” 반응을 보였다(중단 기간 동안 재발 또는 재발되지 않은 환자에서 각각 69.1% [123명/178명], 88.8% [95명/107명]).
위약(시험 I 및 II) 및 MTX(시험 II)와 비교하여 베이스라인으로부터 16주차에 DLQI(피부학적 삶의 질 지수)에서 유의한 개선이 입증되었다. 시험 I에서 SF ‑36의 신체적 및 정신적 구성요소 요약 점수 또한 위약과 비교하여 유의하게 개선되었다.
공개라벨 확장 시험에서 PASI 반응이 50% 미만으로 감소하여 격주 40mg에서 매주 40mg으로 투여량을 증량한 환자 중 각각 26.4% (92명/349명)와 37.8% (132명/349명)는 12주차와 24주차에 PASI 75 반응에 도달하였다.
건선 시험 III (REACH)은 중등증부터 중증의 만성 판상형 건선과 함께 손 및/또는 발 건선이 있는 72명의 환자를 대상으로 위약에 대해 이 약의 효능 및 안전성을 비교하였다. 환자는 이 약 초기 용량 80mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 이 약 40mg을 투여 받거나 위약을 투여 받았다. 16주차에 통계적으로 유의하게 많은 비율의 이 약 투여군 환자가 위약군 환자에 비해 손 및/또는 발에 대해 PGA ‘개선’ 또는 ‘거의 개선’에 도달하였다(각각 30.6% vs. 4.3% [P=0.014]).
건선 시험 IV는 중등증부터 중증의 손발톱 건선이 있는 성인 환자 217명에서 위약에 대해 이 약의 안전성과 유효성을 비교하였다. 환자는 이 약 초기 용량 80mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 이 약 40mg을 투여 받거나 위약을 투여 받았고, 추가 26주 동안 공개라벨 이 약 치료를 받았다. 손발톱 건선은 수정된 손발톱 건선 중증도 지수(mNAPSI) 및 손톱 건선에 대한 의사의 전반적 평가(PGA ‑F)를 사용하여 평가하였다. 위약에 무작위 배정된 환자와 비교하여 이 약에 무작위 배정된 환자 중 통계적으로 유의하게 높은 비율의 환자가 26주차에 mNAPSI에서 최소 75% 개선(mNAPSI 75)에 도달하였다(표 20 참조). NAPSI의 개선율은 이 약 투여군 환자가 위약군 환자보다 16주차(44.2% vs.7.8%)와 26주차(56.2% vs 11.5%)에 통계적으로 유의하게 높았다.
이 약 투여군에서 통계적으로 유의하게 높은 비율의 환자는 위약군과 비교하여 26주차에 베이스라인으로부터 2등급 이상 개선하여 PGA ‑F “개선” 또는 “최소”에 도달하였다. 이 시험에서 이 약은 다양한 범위(BSA ≧10%와 BSA 10% 및 ≧5%)의 손발톱 건선 환자에서 치료 이익을 입증하였으며 위약과 비교하여 두피 건선에서도 통계적으로 유의한 개선을 보였다.
표 20: 26주차의 유효성 결과
|
평가변수 |
위약 N=108 |
격주 이 약 40mg N=109 |
|
≥mNAPSI 75(%) |
3.4 |
46.6a |
|
PGA-F 개선/최소 및 2등급 이상 개선(%) |
6.9 |
48.9a |
|
전체 손발톱 NAPSI 변화율(%) |
-11.5 |
-56.2a |
|
mNAPSI = 0 (%) |
0 |
6.6b |
|
손발톱 통증 수치 등급 척도의 변화 |
-1.1 |
-3.7a |
|
손발톱 건선 신체적 기능 중증도 점수의 변화 |
-0.8 |
-3.7a |
|
B-SNIPI 50 두피(%) |
N=12 0.4 |
N=18 58.3b |
|
a p <0.001, 이 약 vs. 위약 b p <0.05, 이 약 vs. 위약 B-SNIPI 50: 베이스라인 두피 점수보다 6점 이상 증가한 대상자 중 Brigham 두피 손톱 역위 손-발바닥 건선 지수(Brigham Scalp Nail Inverse Palmo-Plantar Psoriasis index; B-SNIPI)의 두피 구성요소가 최소 50% 감소 | ||
52주차까지 이 약 치료를 지속적으로 투여 받은 환자 중 65.0%가 mNAPSI 75 반응에 도달하였고 61.3%는 PGA ‑F 반응에 도달하였다.
이 약을 투여 받은 환자는 위약과 비교하여 26주차의 DLQI(피부학적 삶의 질 지수)가 베이스라인으로부터 통계적으로 유의하게 개선되었다. 26주차에 베이스라인으로부터의 평균 감소(개선)는 이 약군(N=94)에서 8.0, 위약군(N=93)에서 1.9였다.
화농성 한선염
전신 항생제 치료에 불내성 또는 부적절한 반응을 보이거나 금지 사유가 있는 중등증에서 중증의 화농성 한선염(hidradenitis suppurativa; HS) 성인 환자를 대상으로 무작위, 이중눈가림, 위약대조 시험에서 이 약의 안전성과 유효성을 평가하였다. HS ‑I 시험과 HS ‑II 시험의 환자는 최소 3개의 농양 또는 염증성 결절이 있는 Hurley II단계 또는 III단계 질병을 가지고 있었다.
HS ‑I 시험(M11 ‑313)은 2개의 치료 기간에서 307명의 환자를 평가하였다. A 기간에서 환자는 0주차에 위약 또는 이 약 초기 용량 160 mg, 2주차에 80 mg, 4주차부터 11주차까지 매주 40mg을 투여 받았다. 항생제 병용 투여는 시험 동안 허용되지 않았다. 12주간의 치료 후 A 기간에 이 약을 투여 받은 환자는 B 기간에 3개의 치료군 중 하나로 무작위 재배정되었다(12주차부터 35주차까지 매주 이 약 40mg 투여군, 격주 이 약 40mg 투여군, 또는 위약군). A 기간에 위약으로 무작위 배정된 환자는 B 기간에 매주 이 약 40mg 투여군으로 배정하였다.
HS ‑II 시험(M11 ‑810)은 2개의 치료 기간에서 326명의 환자를 평가하였다. A 기간에서 환자는 0주차에 위약 또는 이 약 초기 용량 160 mg, 2주차에 80 mg, 4주차부터 11주차까지 매주 40 mg을 투여 받았다. 환자 중 19.3%는 시험 동안 베이스라인의 경구 항생제 치료를 지속하였다. 12주간의 치료 후 A 기간에 이 약을 투여 받은 환자는 B 기간에 3개의 치료군 중 하나로 무작위 재배정되었다(12주차부터 35주차까지 매주 이 약 40 mg 투여군, 격주 이 약 40 mg 투여군, 또는 위약군). A 기간에 위약으로 무작위 배정된 환자는 B 기간에 위약군으로 배정하였다.
HS ‑I 시험과 HS ‑II 시험에 참여한 환자는 이 약 40 mg을 매주 투여 받는 공개라벨 확장 시험에 등록될 수 있었다. 모든 아달리무맙 투여군의 평균 노출은 762일이었다. 3개의 치료군 모두 국소 소독 세척을 매일 실행하였다.
임상 반응
염증 병변의 임상 반응은 화농성 한선염 임상 반응(HiSCR: 전체 농양 및 염증성 결절 수가 최소 50% 감소하고 베이스라인과 비교하여 농양 수가 증가하거나 배출 누공의 수가 증가하지 않음)을 사용하여 평가하였다. HS와 관련된 피부 통증의 감소는 11점 척도에서 초기 베이스라인 점수가 3점 이상이었던 환자에서 수치 등급 척도를 사용하여 평가하였다.
12주차에 위약과 비교하여 유의하게 높은 비율의 이 약 투여 환자가 HiSCR에 도달하였다. 12주차에 HS ‑II 시험에서 유의하게 높은 비율의 환자는 HS와 관련된 피부 통증이 임상적으로 의미 있게 감소됨을 경험하였다(표 21 참조). 이 약을 투여 받은 환자는 치료 초기 12주 동안 질병 재발의 위험이 유의하게 감소하였다.
표 21: HS 시험 I과 II의 12주차에서의 유효성 결과
|
|
HS 시험 I |
HS 시험 II | ||
|
평가변수 |
위약 |
매주 이 약 40mg |
위약 |
매주 이 약 40mg |
|
화농성 한선염 임상 반응(HiSCR)a |
N=154 40 (26.0%) |
N=153 64 (41.8%)* |
N=163 45 (27.6%) |
N=163 96 (58.9%)*** |
|
피부 통증 ≥30% 감소b |
N=109 27 (24.8%) |
N=122 34 (27.9%) |
N=111 23 (20.7%) |
N=105 48 (45.7%)*** |
|
* p <0.05, 이 약 vs. 위약 *** p <0.001, 이 약 vs. 위약 a 무작위 배정된 모든 환자 b 수치 등급 척도 0-10점을 기초로 베이스라인에서 HS와 관련된 피부 통증 평가 ≥3점인 환자. 0점 = 피부 통증 없음, 10점 = 상상할 수 있는 최악의 피부 통증 | ||||
지속적으로 이 약을 매주 투여 받는 집단에 무작위 배정된 환자의 12주차 전체 HiSCR 비율은 96주차까지 유지되었다. 96주 동안 매주 이 약 40 mg을 장기간 투여 받은 환자에서 새로운 안전성 결과는 확인되지 않았다.
위약과 비교하여 베이스라인으로부터 12주차의 큰 개선은 피부학적 삶의 질 지수(DLQI)로 측정한 피부 특이적 건강 관련 삶의 질(HS ‑I 시험 및 HS ‑II 시험), 치료 만족 설문조사 ‑ 약물(TSQM)로 측정한 환자의 전반적인 약물 치료 만족(HS ‑I 시험 및 HS ‑II 시험), 그리고 SF ‑36의 신체적 구성요소 요약 점수로 측정한 신체적 건강(HS ‑I 시험)으로 입증하였다.
포도막염
독립적인 앞포도막염 환자를 제외하고 비감염성 중간포도막염, 후포도막염 및 전체포도막염(“후부에 영향을 미치는 비감염성 포도막염”으로도 알려짐)이 있는 성인 환자를 대상으로 2건의 무작위, 이중눈가림, 위약대조 시험(UV 시험 I [M10 ‑877] 및 UV 시험 II [M10 ‑880])에서 이 약의 안전성과 유효성을 평가하였다. 환자는 위약을 투여 받거나 초기 용량 80mg의 이 약을 투여 받은 후 일주일 뒤부터 격주로 40mg을 투여 받았다. 안정적 용량의 비생물학적 면역억제제를 병용 투여하는 것은 허용되었다. 두 시험의 일차 효능 평가변수는 ‘치료 실패까지의 시간’이었다. 초기 질병 제어 이후 치료 실패까지의 시간을 연장하면 질병 재발, 염증 및 시력 상실의 위험이 감소된다.
치료 실패는 염증성 맥락망막 및/또는 염증성 망막 혈관 병변, 전방(anterior chamber; AC) 세포 등급, 유리체 혼탁(vitreous haze; VH) 등급 및 최대교정시력(best corrected visual acuity; BCVA)를 기반으로 복합적인 구성요소 결과를 통해 정의하였다.
UV 시험 I은 코르티코스테로이드(10 ‑60mg/일의 경구 프레드니손) 치료에도 불구하고 활동성 포도막염이 있는 환자 217명을 평가하였다. 모든 환자는 시험 등록시 프레드니손의 표준 용량 60mg/일을 투여 받은 후 의무적으로 테이퍼링(tapered) 계획에 따라 코르티코스테로이드를 15주차까지 중단하였다.
UV 시험 II는 베이스라인에서 질병 제어를 위해 만성 코르티코스테로이드(10 ‑35 mg/일의 경구 프레드니손) 치료가 필요한 비활동성 포도막염 환자 226명을 평가하였다. 환자는 이후 의무적으로 테이퍼링 계획에 따라 코르티코스테로이드를 19주차까지 중단하였다.
임상 반응
두 시험 결과는 이 약을 투여 받은 환자가 위약을 투여 받은 환자와 비교하여 치료 실패의 위험이 통계적으로 유의하게 감소했음을 입증하였다(표 22 참조). 두 시험은 위약과 비교하여 치료 실패율에 대한 이 약의 초기에 지속적인 영향을 입증하였다(그림 6 참조).
표 22: UV 시험 I과 II의 치료 실패까지의 시간
|
분석 치료 |
N |
실패 N(%) |
치료 실패까지의 시간(개월) |
HRa |
HRa에 대한 95% CI |
P값b |
|
UV 시험 I에서 6주차 또는 그 이후 치료 실패까지의 시간 | ||||||
|
일차 분석(ITT) | ||||||
|
위약 |
107 |
84 (78.5) |
3.0 |
- |
- |
- |
|
아달리무맙 |
110 |
60 (54.5) |
5.6 |
0.50 |
0.36, 0.70 |
<0.001 |
|
UV 시험 II에서 2주차 또는 그 이후 치료 실패까지의 시간 | ||||||
|
일차 분석(ITT) | ||||||
|
위약 |
111 |
61 (55.0) |
8.3 |
- |
- |
- |
|
아달리무맙 |
115 |
45 (39.1) |
NEc |
0.57 |
0.39, 0.84 |
0.004 |
|
참고: 6주차 이후의 치료 실패(UV 시험 I) 또는 2주차 이후의 치료 실패(UV 시험 II)는 사건으로 계산되었다. 치료 실패 외의 이유로 인한 중단은 중단 시점에서 제외되었다. a 치료를 인자로 설정한 비례 위험 회귀 모델의 아달리무맙 vs. 위약 HR b 로그 순위 검정의 양측 P값 c NE = 추정할 수 없음. 위험 환자의 절반 미만이 사건을 경험하였다. | ||||||
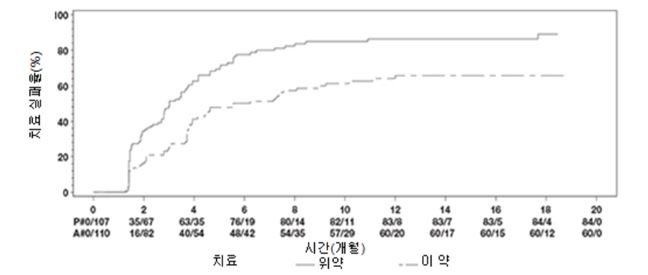
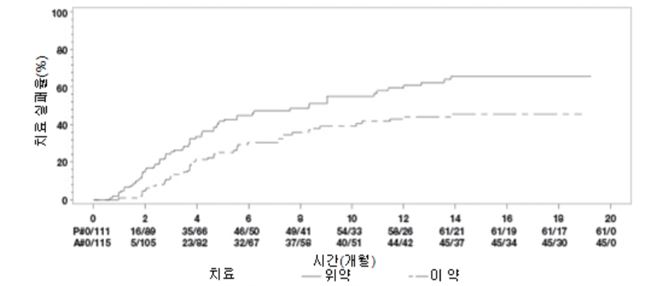
그림 6: 6주차 이후(UV 시험 I) 또는 2주차 이후(UV 시험 II)
치료 실패까지의 시간을 요약한 카플란 ‑메이어 곡선
두 시험 모두에서 일차 평가변수의 모든 요소는 이 약군과 위약군 사이의 전반적인 차이에 점증적으로 기여하였다(표 23).
표 23: UV 시험 I과 II의 치료 실패 구성 요소
|
|
UV I |
UV II | ||||
|
치료 실패까지의 시간의 구성 요소 |
HRa |
95% CI |
P값b |
HRa |
95% CI |
P값b |
|
새로운 활동성 염증성 병변 |
0.38 |
(0.21-0.69) |
0.001 |
0.55 |
(0.26-1.15) |
0.105 |
|
전방 세포 등급 |
0.51 |
(0.30-0.86) |
0.01 |
0.7 |
(0.42-1.18) |
0.18 |
|
유리체 혼탁 등급 |
0.32 |
(0.18-0.58) |
<0.001 |
0.79 |
(0.34-1.81) |
0.569 |
|
최대교정시력 |
0.56 |
(0.32-0.98) |
0.04 |
0.33 |
(0.16-0.70) |
0.002 |
|
참고: 6주차 이후의 치료 실패(UV 시험 I) 또는 2주차 이후의 치료 실패(UV 시험 II)는 사건으로 계산되었다. 치료 실패 외의 이유로 인한 중단은 중단 시점에서 제외되었다. a 치료를 인자로 설정한 비례 위험 회귀 모델의 아달리무맙 vs. 위약 HR b 로그 순위 검정의 양측 P값 | ||||||
또한 UV 시험 I에서 AC 세포 등급, 유리체 혼탁 등급 및 logMAR BCVA(6주차 전의 최적 상태에서 마지막 방문까지의 평균 변화, P값은 각각 0.011, 0.001 및 0.003)의 변화에 대해 위약과 비교하여 아달리무맙에 유리하게 통계적으로 유의한 차이가 관찰되었다.
UV 시험 I 및 UV 시험 II의 비통제 장기간 확장에 포함된 417명의 환자 중 46명이 부적합한 것으로 간주되었고(예: 당뇨망막병증에 의한 합병증 진행, 백내장 수술 또는 유리체절제술로 인해) 효능의 일차 분석에서 제외되었다. 나머지 371명의 환자 중 276명의 평가 가능한 환자는 78주간의 공개라벨 아달리무맙 치료를 완료하였다. 관찰된 데이터 접근법을 기반으로, 222명(80.4%)은 매일 ≦7.5mg의 스테로이드 투여를 동반한 무활동기였고(활동성 염증성 병변 없음, AC 세포 등급 ≦0.5+, VH 등급 ≦0.5+) 184명(66.7%)은 스테로이드를 투여하지 않는 무활동기였다. BCVA는 78주차에 눈의 88.4%에서 개선 또는 유지되었다(5문자 미만으로 하락). 78주 전에 시험을 중단한 환자 중 11%는 이상사례, 5%는 아달리무맙 치료에 대한 불충분한 반응으로 인해 중단하였다.
삶의 질
UV 시험 I에서 이 약 치료는 NEI VFQ ‑25로 측정한 시력과 관련된 기능 및 건강 관련 삶의 질을 유지시켰다.
⓶ 소아
소아 특발성 관절염
다관절성 소아 특발성 관절염(pJIA)
다양한 소아 특발성 관절염 발생 유형(가장 흔한 류마티스인자 음성 또는 양성 다관절성 및 확장성 소수 관절염)을 가진 활동성 다관절성 또는 다관절 과정 소아 특발성 관절염이 있는 소아를 대상으로 2건의 시험(pJIA I 및 II)에서 이 약의 안전성과 유효성을 평가하였다.
pJIA I
다관절성 소아 특발성 관절염(juvenile idiopathic arthritis; JIA)이 있는 171명의 아동(4 ‑ 17세)을 대상으로 다기관, 무작위, 이중눈가림, 평행군 시험에서 이 약의 안전성과 유효성을 평가하였다. 공개라벨 유도 단계(open ‑label lead in; OL LI)에서 환자는 메토트렉세이트를 투여 받은 환자 또는 메토트렉세이트를 투여 받지 않은 환자의 두 집단으로 계층화되었다. 비 ‑메토트렉세이트층에 있는 환자는 메토트렉세이트 투여 이력이 없거나 시험 약물을 투여하기 최소 2주 전에 MTX를 중단한 환자였다. 환자는 안정적 용량의 NSAID 및/또는 프레드니손( ≦0.2 mg/kg/일 또는 최대 10 mg/일)의 투여를 유지하였다. OL LI 단계에서 모든 환자는 24 mg/m2, 최대 40 mg의 이 약을 16주 동안 격주로 투여 받았다. 연령별 환자 분포 및 OL LI 단계 동안 투여 받은 최소, 중앙값, 최대 용량은 표 24에 제시한다.
표 24: 연령별 환자 분포와 OL LI 단계 동안 투여 받은 이 약 용량
|
연령군 |
베이스라인 환자 수 n(%) |
최소, 중앙값 및 최대 용량 |
|
4 - 7세 |
31 (18.1) |
10, 20 및 25mg |
|
8 - 12세 |
71 (41.5) |
20, 25 및 40mg |
|
13 - 17세 |
69 (70.4) |
25, 40 및 40mg |
16주차에 소아 ACR 30 반응을 입증한 환자는 이중눈가림(double blind; OB) 단계에서 무작위 배정되어 추가 32주 동안, 또는 질병이 재발할 때까지 이 약 24 mg/m2, 최대 40 mg을 투여 받거나 위약을 격주로 투여 받았다. 질병 재발 기준은 6개의 소아 ACR 핵심 기준 중 3개 이상이 베이스라인으로부터 ≧30% 악화되고, ≧2개의 활동성 관절 및 6개의 기준 중 >30%의 개선을 보이는 기준이 1개를 넘지 않는 것으로 정의하였다. 32주 후 또는 질병 재발 시 환자는 공개라벨 연장 단계에 등록될 수 있었다.
표 25: JIA 시험에서 소아 ACR 30 반응
|
계층 |
MTX |
비-MTX | ||
|
단계 |
|
| ||
|
OL-LI 16주 |
|
| ||
|
소아 ACR 30 반응(n/N) |
94.1% (80/85) |
74.4% (64/86) | ||
|
| ||||
|
이중눈가림 |
이 약 (n=38) |
위약 (n=37) |
이 약 (n=30) |
위약 (n=28) |
|
32주 마지막에서의 질병 재발a (n/N) |
36.8% (14/38) |
64.9% (24/37)b |
43.3% (13/30) |
71.4% (20/28)c |
|
질병 재발까지의 시간 중앙값 |
>32주 |
20주 |
>32주 |
14주 |
|
a 48주차 소아 ACR 30/50/70 반응은 위약을 투여 받은 환자보다 유의하게 높았다. b p=0.015 c p=0.031 | ||||
16주차에 반응을 보인 환자(n=144) 중, 시험 전반에 걸쳐 이 약을 투여 받은 환자는 OLE 단계에서 최대 6년간 ACR 30/50/90 반응이 유지되었다. 전체적으로 19명의 대상자는 6년 이상 이 약을 투여 받았으며 이들 중 11명은 베이스라인 연령군 4 ‑ 12세, 8명은 베이스라인 연령군 13 ‑ 17세였다.
이 약과 메토트렉세이트를 병용 투여했을 경우 이 약을 단독 투여했을 때보다 전반적인 반응은 일반적으로 우수했으며 보다 적게 항체가 발생하였다. 이러한 결과를 고려해볼 때, 이 약을 메토트렉세이트와 병용 투여하는 것이 권장되며 메토트렉세이트의 투여가 적절하지 않은 환자는 단일 요법으로 사용하는 것이 권장된다.
pJIA II
중등증부터 중증의 활동성 다관절성 JIA가 있는 32명의 아동(2세 ‑4세 아동 또는 체중이 15kg인 4세 이상의 아동)을 대상으로 공개라벨, 다기관 시험에서 이 약의 안전성과 유효성을 평가하였다. 환자는 24 mg/m2 체표면적(body surface area; BSA), 최대 20 mg의 이 약을 최소 24주 동안 SC 단일 주입을 통해 격주로 투여 받았다. 시험 동안 대부분의 대상자는 MTX를 병용 투여하고 있었으며 일부에서 코르티코스테로이드 또는 NSAID 병용 투여를 보고하였다.
12주차와 24주차에 관찰된 데이터 접근법을 사용한 소아 ACR 30 반응은 각각 93.5%와 90.0%였다. 12주차와 24주차에 소아 ACR 50/70/90에 도달한 대상자의 비율은 각각 90.3%/61.3%/38.7%와 83.3%/73.3%/36.7%였다. 24주차에 반응(소아 ACR 30)을 보인 환자(30명 중 27명) 중 이 기간 동안 이 약을 투여 받은 환자는 OLE 단계에서 최대 60주 동안 ACR 30 반응이 유지되었다. 전반적으로 20명의 대상자가 60주 이상 이 약을 투여 받았다.
골부착부염 관련 관절염
골부착부염 관련 관절염이 있는 46명의 소아 환자(6 ‑ 17세)를 대상으로 다기관, 무작위, 이중눈가림 시험에서 이 약의 안전성과 유효성을 평가하였다. 환자는 24 mg/m2 체표면적(body surface area; BSA), 최대 40 mg의 이 약 또는 위약을 12주 동안 투여하는 집단 중 하나로 무작위 배정되었다. 이중눈가림 기간 후 공개라벨(open ‑label; OL) 기간 동안 환자는 이 약 24 mg/m2 BSA, 최대 40 mg을 추가 192주 동안 격주로 피하 투여 받았다. 일차 평가변수는 베이스라인부터 12주차까지 관절염(기형 또는 통증 및/또는 압통을 동반한 운동 상실 관절로 인한 것이 아닌 부종)이 있는 활동성 관절 수의 변화율이었으며 이 약군의 평균 비율 감소는 ‑62.6%인 반면 위약군에서는 ‑11.6%였다. 관절염이 있는 활동성 관절 수의 개선은 공개라벨 기간 동안 156주차까지 유지되었다. 환자의 대다수는 부착부염 부위의 수, 압통 관절 수(tender joint count; TJC), 종창 관절 수(swollen joint count; SJC), 소아 ACR 30 반응, 소아 ACR 50 반응 및 소아 ACR 70 반응과 같은 이차 평가변수의 임상적 개선을 입증하였으며, 이러한 개선은 공개라벨 기간 동안 156주차까지 유지되었다.
소아 크론병
소아 크론병 활동도 지수(Pediatric Crohn's Disease Activity Index; PCDAI) 점수가 >30인 것으로 정의되는 중등증부터 중증의 크론병(Crohn’s disease; CD)이 있는 6 ‑ 17세의 소아 환자 192명을 대상으로 체중(40 kg 또는 ≧40 kg)에 따른 이 약의 유도 및 유지 치료에 대한 효능 및 안전성을 평가하기 위해 다기관, 무작위, 이중눈가림 임상시험을 설계하여 이 약을 평가하였다. 대상자는 CD에 대한 기존 치료(코르티코스테로이드 및/또는 면역조절제 포함)에 실패한 이력이 있어야 했다. 또한 대상자는 인플릭시맙에 대한 반응 도달에 실패했거나 불내성이 있을 수 있다.
모든 대상자는 베이스라인 체중을 기반으로 한 투여량으로 공개라벨 유도 치료를 받았다. 체중이 ≧40 kg인 환자의 경우 0주차에 160 mg, 2주차에 80 mg을 투여 받았으며 40 kg인 환자의 경우에는 각각 80 mg과 40 mg을 투여 받았다.
4주차에 대상자는 표 26에 제시된 바와 같이 저용량 또는 표준 용량 유지 요법을 받는 시점의 체중에 따라 각 투여군에 1:1로 무작위 배정되었다.
표 26: 유지 요법
|
환자 체중 |
저용량 |
표준용량 |
|
<40kg |
격주 10mg |
격주 20mg |
|
≥40kg |
격주 20mg |
격주 40mg |
유효성 결과
시험의 일차 평가변수는 26주차에 PCDAI 점수 ≦10점으로 정의되는 임상적 관해였다.
임상적 관해 및 임상 반응(PCDAI 점수가 베이스라인으로부터 최소 15점 감소되는 것으로 정의) 비율은 표 27에 제시한다. 코르티코스테로이드 또는 면역조절제의 중단율은 표 28에 제시한다.
표 27: 소아 CD 시험의 PCDAI 임상적 관해 및 반응
|
|
표준용량 격주 40/20mg N=93 |
저용량 격주 20/10mg N=95 |
P값* |
|
26주차 |
|
|
|
|
임상적 관해 |
38.7% |
28.4% |
0.075 |
|
임상 반응 |
59.1% |
48.4% |
0.073 |
|
52주차 |
|
|
|
|
임상적 관해 |
33.3% |
23.2% |
0.100 |
|
임상 반응 |
41.9% |
28.4% |
0.038 |
|
* 표준용량 vs 저용량 비교에 대한 p값 | |||
표 28: 소아 CD 시험의 코르티코스테로이드 또는 면역조절제 중단 및 누공 관해
|
|
표준용량 격주 40/20mg |
저용량 격주 20/10mg |
P값1 |
|
코르티코스테로이드 중단 |
N=33 |
N=38 |
|
|
26주차 |
84.8% |
65.8% |
0.066 |
|
52주차 |
69.7% |
60.5% |
0.420 |
|
면역조절제 중단2 |
N=60 |
N=57 |
|
|
52주차 |
30.0% |
29.8% |
0.983 |
|
누공 관해3 |
N=15 |
N=21 |
|
|
26주차 |
46.7% |
38.1% |
0.608 |
|
52주차 |
40.0% |
23.8% |
0.303 |
|
1 표준용량 vs 저용량 비교에 대한 p값 2 면역조절제 치료는 대상자가 임상 반응 기준을 충족했을 때 시험자의 재량으로 26주차 이후에만 중단할 수 있었다. 3 최소 2번의 연속적인 베이스라인 후 방문 동안, 베이스라인에서 배출이 진행되었던 모든 누공의 폐쇄로 정의 | |||
26주차와 52주차에 두 치료군에서 체질량지수 및 신장 속도에 대해 베이스라인으로부터 통계학적으로 유의한 증가(개선)가 관찰되었다. 삶의 질 매개변수(IMPACT III 포함)에 대해서도 두 치료군 모두에서 베이스라인으로부터 통계적 및 임상적으로 유의한 개선이 관찰되었다.
소아 CD 시험의 환자는 공개라벨 장기간 확장 시험에서 투여를 지속할 수 있는 선택권이 있었다. 5년간의 이 약 치료 이후 환자의 74% (37명/50명)은 임상적 관해가 지속되었고 환자의 92% (46명/50명)는 PCDAI에 따라 임상 반응이 지속되었다.
소아 판상 건선
국소 치료 및 일광요법이나 광선요법으로 부적절하게 제어된 중증의 만성 판상형 건선(PGA ≧4 또는 >20% BSA 관련 또는 매우 두꺼운 병변을 동반한 >10 % BSA 관련 또는 PASI ≧20 또는 임상적으로 관련된 얼굴, 생식기, 손/발 병변을 동반한 PASI ≧10으로 정의)이 있는 4세 소아 환자 114명을 대상으로 무작위, 이중눈가림, 통제 시험에서 이 약의 효능을 평가하였다.
환자는 격주 0.8 mg/kg(최대 40mg) 이 약, 격주 0.4 mg/kg(최대 20mg) 이 약, 또는 매주 0.1 ‑0.4 mg/kg(최대 25mg) 메토트렉세이트를 투여 받았다. 16주차에 이 약 0.8 mg/kg 투여군에 무작위 배정된 환자가 MTX에 무작위 배정된 환자보다 더 많은 수에서 호의적인 유효성 반응(예: PASI 75)을 보였다.
표 29: 16주차의 소아 판상 건선 유효성 결과
|
|
MTXa N=37 |
이 약 격주 0.8mg/kg N=38 |
|
PASI 75b |
12 (32.4%) |
22 (57.9%) |
|
PGA: 개선/최소c |
15 (40.5%) |
23 (60.5%) |
|
a MTX = 메토트렉세이트 b P=0.027, 이 약 0.8mg/kg vs. MTX c P=0.083, 이 약 0.8mg/kg vs. MTX | ||
PASI 75 및 PGA 개선이나 최소에 도달한 환자는 최대 36주간의 치료를 중단하였으며 질병 제어의 실패(PGA 반응 도달 실패)에 대해 모니터링되었다. 그런 다음 환자는 추가 16주 동안 아달리무맙 0.8 mg/kg을 격주로 재투여하였으며 재투여 동안 관찰된 반응은 이전의 이중눈가림 기간에 관찰된 것과 유사하였다: PASI 75 반응 78.9%(19명 중 15명), PGA 개선 또는 최소 52.6 %(19명 중 10명).
시험의 공개라벨 기간에 PASI 75 및 PGA 개선이나 최소 반응은 새로운 안전성 결과 없이 추가적으로 최대 52주 동안 유지되었다.
⓷ 면역원성
항 ‑아달리무맙 항체의 형성은 아달리무맙의 청소율 증가 및 효능 감소와 관련된다. 항 ‑아달리무맙 항체의 존재와 이상반응 사이의 상관관계는 명백하지 않다.
면역원성 분석이 의약품마다 다르기 때문에 다른 의약품의 항체 비율과 비교하는 것은 오해의 소지가 있을 수 있다.
성인
류마티스 관절염 시험 I, II 및 III의 환자에서 6 ‑12개월 동안 다양한 시점에서 항 ‑아달리무맙 항체에 대해 검사하였다. 확증시험에서 항 ‑아달리무맙 항체가 아달리무맙을 투여 받은 환자의 5.5% (58명/1053명)에서 확인되었고, 그에 반해 위약을 투여 받은 환자에서는 0.5% (2명/370명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 12.4%였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 0.6 %였다.
건선성 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 10 % (38명/376명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 13.5 % (24명/178명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 7 % (14명/198명)였다.
강직성 척추염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 8.3 % (17명/204명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 8.6 % (16명/185명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 5.3 % (1명/19명)였다.
비방사선학적 축성 척추관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 지속적으로 투여 받은 환자의 8명/152명(5.3 %)에서 확인되었다.
크론병 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 2.6% (7명/269명)에서 확인되었다.
중등증에서 중증의 활동성 UC 환자에서 아달리무맙을 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 5.0%였다.
장내 베체트병이 있는 일본인 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 5% (1명/20명)에서 확인되었다.
건선 환자에서 항 ‑아달리무맙 항체는 메토트렉세이트를 병용 투여하지 않은 아달리무맙 투여 환자의 8.4 % (77명/920명)에서 확인되었다. 메토트렉세이트를 병용 투여하지 않고 장기간 아달리무맙을 투여하고 중단 및 재투여 시험에 참여한 판상형 건선 환자에서 재투여 후 항 ‑아달리무맙 항체의 발생률은 2.3 %였고 중단 전에 관찰된 발생률인 1.9%와 유사하였다.
중등증에서 중증의 화농성 한선염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 10.1% (10명/99명)에서 확인되었다.
비감염성 포도막염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 4.8% (12명/249명)에서 확인되었다.
소아
4 ‑ 17세의 다관절성 소아 특발성 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 16%에서 확인되었다. 메토트렉세이트를 병용하지 않은 환자의 경우 발생률은 26%였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 6%였다. 2세부터 4세 또는 체중이 15 kg인 4세 이상의 다관절성 소아 특발성 관절염 환자의 7%에서 항 ‑아달리무맙 항체가 확인되었고 1명의 환자는 메토트렉세이트를 병용 투여하는 중이었다.
골부착부위염 관련 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 11% (5명/46명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 14% (3명/22명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 8% (2명/24명)였다.
중등증에서 중증의 활동성 소아 크론병 환자에서 아달리무맙을 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 3.3%였다.
소아 건선 환자에서 0.8 mg/kg 아달리무맙을 단일 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 13% (5명/38명)였다.
3) 비임상 안전성 자료
비임상 데이터에 따르면 아달리무맙은 단회 투여 독성, 반복 투여 독성 및 유전 독성 시험을 근거로 인간에 대해 특별한 위해성을 나타내지 않는다.
배아 태아 발달 독성/주산기 발달 연구는 사이노몰로거스(cynomologous) 원숭이에서 0, 30 및 100 mg / kg (그룹 당 9 ‑17 마리의 원숭이)의 용량으로 수행되었으며, 아달리무맙이 태아에게 해를 입힌다는 증거는 없었다. 설치류 TNF에 대한 교차 반응이 제한적인 항체 및 설치류에서 중화 항체의 개발에 대한 적절한 모델이 없기 때문에 아달리무맙에 대한 발암성 연구나 생식기능 및 출생 후 독성에 대한 표준 평가는 수행되지 않았다.
성인(18세 이상)
1. 류마티스 관절염
메토트렉세이트를 포함한 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대한 반응이 적절하지 않은 성인의 중등도에서 중증의 활동성 류마티스 관절염의 치료
이전에 메토트렉세이트로 치료받지 않은 성인의 중증의 활동성 및 진행성 류마티스 관절염의 치료
이 약은 단독투여 또는 메토트렉세이트나 다른 DMARDs와 병용투여할 수 있다. 메토트렉세이트에 내약성이 없거나 지속적인 병용투여가 부적절한 경우에는 단독투여한다.
이 약과 메토트렉세이트와의 병용투여는 관절손상 진행속도를 감소시키고(X ‑선측정 결과) 신체활동기능을 향상시킨다.
2. 건선성 관절염
이전에 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대한 반응이 적절하지 않은 성인의 활동성 및 진행성 건선성 관절염의 치료. 이 약은 다발성, 대칭성 아형을 지닌 환자의 말초 관절손상 진행속도를 감소시키고(X ‑선측정 결과), 신체활동기능을 향상시킨다.
3. 축성 척추관절염
(1) 강직성 척추염
기존 치료에 대한 반응이 적절하지 않은 성인의 중증의 강직성 척추염의 치료
(2) 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염
방사선학적 검사에서 강직성 척추염이 확인되지 않으나, 상승된 CRP 수치 및/또는 MRI상 객관적인 염증의 징후를 보이는 중증 축성 척추관절염의 치료. 이 약은 비스테로이드성항염증제(NSAIDs) 약물에 대한 반응이 적절하지 않거나, 불내성인 환자에 사용한다.
4. 크론병
코르티코스테로이드제나 면역억제제 등의 치료에 반응을 나타내지 않거나, 내약성이 없는 경우 또는 이러한 치료방법이 금기인 중등도에서 중증의 활성 크론병의 치료
유도 요법의 경우 이 약은 코르티코스테로이드제와 병용투여한다. 코르티코스테로이드제에 내약성이 없거나, 지속적인 병용투여가 부적절한 경우 단독투여할 수 있다.
5. 건선
전신치료 또는 광선요법이 필요한 성인의 중등도에서 중증의 만성 판상 건선의 치료
6. 궤양성 대장염
코르티코스테로이드 및 6 ‑MP(6 ‑mercaptopurine) 또는 AZA(azathioprine)를 포함한 통상적인 치료에 대해 반응이 적절하지 않거나 내약성이 없는 경우 또는 이러한 치료방법이 금기인 성인의 중등도에서 중증의 활성 궤양성 대장염의 치료
7. 베체트 장염
스테로이드 또는 면역억제제 등의 통상적인 치료에도 적절한 반응이 나타나지 않는 베체트 장염의 치료
8. 화농성 한선염
기존의 전신 요법에 적절한 반응을 나타내지 않는 중등도에서 중증의 활성 화농성 한선염의 치료
9. 포도막염
코르티코스테로이드에 적절한 반응을 나타내지 않은 성인의 비 ‑감염성 중간 포도막염, 후포도막염 및 전체포도막염의 치료
소아
1. 소아 크론병(6 ‑ 17세)
일차 영양요법, 코르티코스테로이드, 면역조절제 등의 치료에 적절한 반응을 나타내지 않거나, 내약성이 없는 경우 또는 이러한 치료방법이 금기인 소아 환자(6 ‑ 17세)에서 중등도에서 중증의 활성 크론병의 치료
2. 소아 특발성 관절염
(1) 다관절형 소아 특발성 관절염
하나 이상의 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대해 부적절한 반응을 보인 2세 이상의 소아 및 청소년에서 활성 다관절형 소아 특발성 관절염의 치료
(2) 골부착부위염 관련 관절염
기존 치료에 적절한 반응을 나타내지 않거나 내약성이 없는 6세 이상 어린이 및 청소년의 활성 골부착부위염 관련 관절염의 치료
3. 소아 판상 건선
국소 치료 및 광선요법에 적절한 반응을 나타내지 않거나 해당 치료가 부적절한 4세 이상의 어린이 및 청소년의 중증 만성 판상 건선의 치료
1. 류마티스 관절염
메토트렉세이트를 포함한 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대한 반응이 적절하지 않은 성인의 중등도에서 중증의 활동성 류마티스 관절염의 치료
이전에 메토트렉세이트로 치료받지 않은 성인의 중증의 활동성 및 진행성 류마티스 관절염의 치료
이 약은 단독투여 또는 메토트렉세이트나 다른 DMARDs와 병용투여할 수 있다. 메토트렉세이트에 내약성이 없거나 지속적인 병용투여가 부적절한 경우에는 단독투여한다.
이 약과 메토트렉세이트와의 병용투여는 관절손상 진행속도를 감소시키고(X ‑선측정 결과) 신체활동기능을 향상시킨다.
2. 건선성 관절염
이전에 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대한 반응이 적절하지 않은 성인의 활동성 및 진행성 건선성 관절염의 치료. 이 약은 다발성, 대칭성 아형을 지닌 환자의 말초 관절손상 진행속도를 감소시키고(X ‑선측정 결과), 신체활동기능을 향상시킨다.
3. 축성 척추관절염
(1) 강직성 척추염
기존 치료에 대한 반응이 적절하지 않은 성인의 중증의 강직성 척추염의 치료
(2) 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염
방사선학적 검사에서 강직성 척추염이 확인되지 않으나, 상승된 CRP 수치 및/또는 MRI상 객관적인 염증의 징후를 보이는 중증 축성 척추관절염의 치료. 이 약은 비스테로이드성항염증제(NSAIDs) 약물에 대한 반응이 적절하지 않거나, 불내성인 환자에 사용한다.
4. 크론병
코르티코스테로이드제나 면역억제제 등의 치료에 반응을 나타내지 않거나, 내약성이 없는 경우 또는 이러한 치료방법이 금기인 중등도에서 중증의 활성 크론병의 치료
유도 요법의 경우 이 약은 코르티코스테로이드제와 병용투여한다. 코르티코스테로이드제에 내약성이 없거나, 지속적인 병용투여가 부적절한 경우 단독투여할 수 있다.
5. 건선
전신치료 또는 광선요법이 필요한 성인의 중등도에서 중증의 만성 판상 건선의 치료
6. 궤양성 대장염
코르티코스테로이드 및 6 ‑MP(6 ‑mercaptopurine) 또는 AZA(azathioprine)를 포함한 통상적인 치료에 대해 반응이 적절하지 않거나 내약성이 없는 경우 또는 이러한 치료방법이 금기인 성인의 중등도에서 중증의 활성 궤양성 대장염의 치료
7. 베체트 장염
스테로이드 또는 면역억제제 등의 통상적인 치료에도 적절한 반응이 나타나지 않는 베체트 장염의 치료
8. 화농성 한선염
기존의 전신 요법에 적절한 반응을 나타내지 않는 중등도에서 중증의 활성 화농성 한선염의 치료
9. 포도막염
코르티코스테로이드에 적절한 반응을 나타내지 않은 성인의 비 ‑감염성 중간 포도막염, 후포도막염 및 전체포도막염의 치료
소아
1. 소아 크론병(6 ‑ 17세)
일차 영양요법, 코르티코스테로이드, 면역조절제 등의 치료에 적절한 반응을 나타내지 않거나, 내약성이 없는 경우 또는 이러한 치료방법이 금기인 소아 환자(6 ‑ 17세)에서 중등도에서 중증의 활성 크론병의 치료
2. 소아 특발성 관절염
(1) 다관절형 소아 특발성 관절염
하나 이상의 DMARDs(disease ‑modifying anti ‑rheumatic drugs)에 대해 부적절한 반응을 보인 2세 이상의 소아 및 청소년에서 활성 다관절형 소아 특발성 관절염의 치료
(2) 골부착부위염 관련 관절염
기존 치료에 적절한 반응을 나타내지 않거나 내약성이 없는 6세 이상 어린이 및 청소년의 활성 골부착부위염 관련 관절염의 치료
3. 소아 판상 건선
국소 치료 및 광선요법에 적절한 반응을 나타내지 않거나 해당 치료가 부적절한 4세 이상의 어린이 및 청소년의 중증 만성 판상 건선의 치료
환자가 자가 주사하는 것이 적절하며 필요시 치료 추적이 가능하다고 의사가 판단하는 경우, 환자는 주사방법에 대한 교육을 받은 후 이 약을 자가 주사할 수 있다.
80 mg의 투여는 하루에 40 mg을 2번 주사한다. 160 mg의 투여는 하루에 40 mg을 4번 주사하거나, 이틀에 걸쳐 40 mg을 2번씩 주사할 수 있다.
성인
1. 류마티스 관절염
성인 류마티스 관절염 환자에 대한 이 약의 권장 용량은 아달리무맙 40 mg을 2주에 1회 피하주사한다. 이 약을 투여하는 동안 메토트렉세이트의 병용투여를 유지한다.
글루코코르티코이드, 살리실산염, 비스테로이드성항염증제(NSAIDs), 진통제, 다른 DMARDs의 병용투여를 유지할 수 있다.
단독요법의 경우 이 약에 대한 반응이 감소한 환자는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하면 유용한 효과를 얻을 수 있다.
2. 건선성 관절염, 강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염
건선성 관절염, 강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염 환자에 대한 이 약의 권장 용량은 아달리무맙 40 mg을 2주에 1회 피하주사한다.
상기 효능 ▪효과에 대해 임상적인 반응은 보통 치료 12주 이내에 나타나며, 이 기간 내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
3. 성인 크론병
성인 중등도에서 중증 크론병에 대한 이 약의 권장 용량은 첫 주에 아달리무맙 80 mg을 투여하고 첫 투여 후 2주 후에 40 mg을 투여한다. 빠른 효과를 얻어야 할 필요가 있는 경우에는, 유도 요법동안 이상 반응에 대한 위험성이 증가한다는 것을 알리고, 첫 주에 160 mg을 투여하고 첫 투여 후 2주 후에 80 mg을 투여할 수 있다. 유도 요법의 경우 이 약은 코르티코스테로이드제와 병용투여한다. 코르티코스테로이드제에 내약성이 없거나, 지속적인 병용투여가 부적절한 경우 단독투여할 수 있다.
유도 요법 후, 아달리무맙 40 mg을 2주에 1회 피하주사한다. 만약 투여를 중지하고 재발의 증상과 징후가 나타나면 이 약을 재투여 할 수 있다. 8주 이상의 투여 중지 후 재투여에 대한 경험은 거의 없다.
유지 요법 동안 임상 지침에 따라 코르티코스테로이드제의 투여를 서서히 줄일 수 있다.
이 약에 대한 반응이 감소한 환자는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하면 유용한 효과를 얻을 수 있다.
4주까지 반응을 보이지 않는 환자의 경우 12주까지 투여하여 반응을 나타낼 수 있다. 이 기간 내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
4. 건선
성인에 대한 이 약의 권장 용량은 첫 회에 아달리무맙 80 mg을 피하로 투여하고, 이어서 첫 투여 후 1주일 후에 40 mg을 격주로 투여한다.
16주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
충분한 반응이 나타나지 않는 환자는 40mg 매주 투여 또는 80 mg 격주 투여로 증량하여 유용한 효과를 얻을 수 있다. 증량 후 충분한 반응이 나타나지 않는 환자의 경우에는 이 약의 지속적 40 mg 매주 투여 또는 80 mg 격주 투여의 유용성과 위험성을 신중히 재고한다.
5. 궤양성 대장염
성인 중등도에서 중증의 궤양성 대장염에 대한 이 약의 권장 용량은 아달리무맙 160 mg을 투여하고 첫 투여 2주 후에 80 mg을 투여하는 것이다. 유도 요법 후 40 mg을 격주로 피하주사한다.
유지 요법 동안 임상 지침에 따라 코르티코스테로이드제의 투여를 서서히 줄일 수 있다.
이 약에 대한 반응이 감소한 환자 중 일부는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하여 유용한 효과를 얻을 수 있다.
지금까지 나온 데이터에 따르면 임상 반응은 대체로 투여 2 ‑8주 이내에 도달한다. 이 기간 내에 반응을 보이지 않은 환자의 경우 투여 지속여부를 신중히 재고한다.
6. 베체트 장염
성인에 대한 이 약의 권장 용량은 첫 회에 160 mg을 피하로 투여하고 이어서 첫 투여 2주 후에 80 mg을 투여한다. 첫 투여 4주 후부터는 40 mg을 격주로 투여한다.
12주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
7. 화농성 한선염
이 약의 권장용량은 첫 회에 160 mg을 피하로 투여하고, 이어서 첫 투여 2주 후에 80 mg을 투여한다. 첫 투여 4 주 후 부터는 40 mg을 매주 투여 또는 80 mg을 격주 투여한다. 필요 시, 이 약의 치료 기간 동안 항생제를 계속 투여할 수 있다.
12주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
치료를 중단해야 하는 경우, 40mg 매주 투여 또는 80 mg 격주 투여로 재투여할 수 있다.
8. 포도막염
성인에 대한 이 약의 권장 용량은 첫 회 80 mg 투여하고 이어서 첫 투여 1주 후부터 40mg을 격주로 투여한다.
이 약은 단독 또는 코르티코스테로이드 또는 면역억제제 (생물학적 면역억제제 제외)와 병용하여 사용할 수 있다. 코르티코스테로이드는 임상 경험에 따라 양을 줄일 수 있다.
소아
1. 소아 크론병
6세 이상의 소아 크론병 환자에서 이 약의 권장 투여 용량은 체중을 기반으로 한다. 이 약은 피하 투여한다.
충분한 반응이 나타나지 않는 환자의 경우 증량하여 유용한 효과를 얻을 수 있다.
◦ 40 kg 미만: 20 mg 매주 투여
◦ 40 kg 이상: 40 mg 매주 투여 또는 80 mg 격주 투여
12주까지 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
2. 소아 특발성 관절염
(1) 다관절형 소아 특발성 관절염
이 약은 2세 미만 소아 또는 체중 10 kg 미만 환자를 대상으로 연구된 바 없다.
이 약의 권장 투여 용량은 체중을 기반으로 한다. 이 약은 메토트렉세이트와 병용하여 투여한다. 메토트렉세이트에 내약성이 없거나 메토트렉세이트와 함께 지속적으로 투여하는 것이 부적절할 경우에는 이 약을 단독요법으로 투여할 수 있다.
지금까지 나온 데이터에 따르면 임상 반응은 대체로 투여 12주 이내에 도달한다. 이 기간 내에 반응을 보이지 않은 환자의 경우 투여 지속여부를 신중히 재고한다.
(2) 골부착부위염 관련 관절염
6세 이상의 골부착부위염 관련 관절염 환자에 대한 이 약의 권장 투여 용량은 체중을 기반으로 한다.
이 약은 6세 미만의 골부착부위염 관련 관절염 환자를 대상으로 연구된 바 없다.
3. 소아 판상 건선
소아 판상 건선에 대한 이 약의 권장 투여 용량은 체중을 기반으로 한다. 16주 이후에도 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
만약 이 약으로 다시 치료할 경우, 위에서 제시한 용량과 치료기간을 따라야 한다.
소아 판상 건선으로 4세 미만의 어린이에게 이 약을 사용한 적은 없다.
80 mg의 투여는 하루에 40 mg을 2번 주사한다. 160 mg의 투여는 하루에 40 mg을 4번 주사하거나, 이틀에 걸쳐 40 mg을 2번씩 주사할 수 있다.
성인
1. 류마티스 관절염
성인 류마티스 관절염 환자에 대한 이 약의 권장 용량은 아달리무맙 40 mg을 2주에 1회 피하주사한다. 이 약을 투여하는 동안 메토트렉세이트의 병용투여를 유지한다.
글루코코르티코이드, 살리실산염, 비스테로이드성항염증제(NSAIDs), 진통제, 다른 DMARDs의 병용투여를 유지할 수 있다.
단독요법의 경우 이 약에 대한 반응이 감소한 환자는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하면 유용한 효과를 얻을 수 있다.
2. 건선성 관절염, 강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염
건선성 관절염, 강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염 환자에 대한 이 약의 권장 용량은 아달리무맙 40 mg을 2주에 1회 피하주사한다.
상기 효능 ▪효과에 대해 임상적인 반응은 보통 치료 12주 이내에 나타나며, 이 기간 내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
3. 성인 크론병
성인 중등도에서 중증 크론병에 대한 이 약의 권장 용량은 첫 주에 아달리무맙 80 mg을 투여하고 첫 투여 후 2주 후에 40 mg을 투여한다. 빠른 효과를 얻어야 할 필요가 있는 경우에는, 유도 요법동안 이상 반응에 대한 위험성이 증가한다는 것을 알리고, 첫 주에 160 mg을 투여하고 첫 투여 후 2주 후에 80 mg을 투여할 수 있다. 유도 요법의 경우 이 약은 코르티코스테로이드제와 병용투여한다. 코르티코스테로이드제에 내약성이 없거나, 지속적인 병용투여가 부적절한 경우 단독투여할 수 있다.
유도 요법 후, 아달리무맙 40 mg을 2주에 1회 피하주사한다. 만약 투여를 중지하고 재발의 증상과 징후가 나타나면 이 약을 재투여 할 수 있다. 8주 이상의 투여 중지 후 재투여에 대한 경험은 거의 없다.
유지 요법 동안 임상 지침에 따라 코르티코스테로이드제의 투여를 서서히 줄일 수 있다.
이 약에 대한 반응이 감소한 환자는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하면 유용한 효과를 얻을 수 있다.
4주까지 반응을 보이지 않는 환자의 경우 12주까지 투여하여 반응을 나타낼 수 있다. 이 기간 내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
4. 건선
성인에 대한 이 약의 권장 용량은 첫 회에 아달리무맙 80 mg을 피하로 투여하고, 이어서 첫 투여 후 1주일 후에 40 mg을 격주로 투여한다.
16주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
충분한 반응이 나타나지 않는 환자는 40mg 매주 투여 또는 80 mg 격주 투여로 증량하여 유용한 효과를 얻을 수 있다. 증량 후 충분한 반응이 나타나지 않는 환자의 경우에는 이 약의 지속적 40 mg 매주 투여 또는 80 mg 격주 투여의 유용성과 위험성을 신중히 재고한다.
5. 궤양성 대장염
성인 중등도에서 중증의 궤양성 대장염에 대한 이 약의 권장 용량은 아달리무맙 160 mg을 투여하고 첫 투여 2주 후에 80 mg을 투여하는 것이다. 유도 요법 후 40 mg을 격주로 피하주사한다.
유지 요법 동안 임상 지침에 따라 코르티코스테로이드제의 투여를 서서히 줄일 수 있다.
이 약에 대한 반응이 감소한 환자 중 일부는 40 mg 매주 투여 또는 80 mg 격주 투여로 증량하여 유용한 효과를 얻을 수 있다.
지금까지 나온 데이터에 따르면 임상 반응은 대체로 투여 2 ‑8주 이내에 도달한다. 이 기간 내에 반응을 보이지 않은 환자의 경우 투여 지속여부를 신중히 재고한다.
6. 베체트 장염
성인에 대한 이 약의 권장 용량은 첫 회에 160 mg을 피하로 투여하고 이어서 첫 투여 2주 후에 80 mg을 투여한다. 첫 투여 4주 후부터는 40 mg을 격주로 투여한다.
12주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
7. 화농성 한선염
이 약의 권장용량은 첫 회에 160 mg을 피하로 투여하고, 이어서 첫 투여 2주 후에 80 mg을 투여한다. 첫 투여 4 주 후 부터는 40 mg을 매주 투여 또는 80 mg을 격주 투여한다. 필요 시, 이 약의 치료 기간 동안 항생제를 계속 투여할 수 있다.
12주 이내에 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
치료를 중단해야 하는 경우, 40mg 매주 투여 또는 80 mg 격주 투여로 재투여할 수 있다.
8. 포도막염
성인에 대한 이 약의 권장 용량은 첫 회 80 mg 투여하고 이어서 첫 투여 1주 후부터 40mg을 격주로 투여한다.
이 약은 단독 또는 코르티코스테로이드 또는 면역억제제 (생물학적 면역억제제 제외)와 병용하여 사용할 수 있다. 코르티코스테로이드는 임상 경험에 따라 양을 줄일 수 있다.
소아
1. 소아 크론병
6세 이상의 소아 크론병 환자에서 이 약의 권장 투여 용량은 체중을 기반으로 한다. 이 약은 피하 투여한다.
|
체중 (kg) |
유도 용량 |
유지 용량 |
|
40 kg 미만 |
ㆍ 첫 주에 40mg 투여하고 첫 투여 후 2주 후에 20mg 투여 ㆍ 빠른 효과를 얻어야 할 필요가 있는 경우에는, 고용량의 유도요법 동안 이상사례에 대한 위험성이 증가한다는 것을 알리고 아래 용량을 투여할 수 있다. - 첫 주에 80mg 투여하고, 첫 투여 후 2주 후에 40mg 투여 |
20mg 격주 투여 |
|
40 kg 이상 |
ㆍ 첫 주에 80mg 투여하고 첫 투여 후 2주 후에 40mg 투여 ㆍ 빠른 효과를 얻어야 할 필요가 있는 경우에는, 고용량의 유도요법 동안 이상사례에 대한 위험성이 증가한다는 것을 알리고 아래 용량을 투여할 수 있다. - 첫 주에 160mg 투여하고, 첫 투여 후 2주 후에 80mg 투여 |
40mg 격주 투여 |
◦ 40 kg 미만: 20 mg 매주 투여
◦ 40 kg 이상: 40 mg 매주 투여 또는 80 mg 격주 투여
12주까지 반응을 나타내지 않는 환자의 경우에는 투여 지속 여부를 신중히 재고한다.
2. 소아 특발성 관절염
(1) 다관절형 소아 특발성 관절염
이 약은 2세 미만 소아 또는 체중 10 kg 미만 환자를 대상으로 연구된 바 없다.
이 약의 권장 투여 용량은 체중을 기반으로 한다. 이 약은 메토트렉세이트와 병용하여 투여한다. 메토트렉세이트에 내약성이 없거나 메토트렉세이트와 함께 지속적으로 투여하는 것이 부적절할 경우에는 이 약을 단독요법으로 투여할 수 있다.
|
체중 (kg) |
용량 |
|
10 kg 이상 30 kg 미만 |
20mg 격주 투여 |
|
30 kg 이상 |
40mg 격주 투여 |
(2) 골부착부위염 관련 관절염
6세 이상의 골부착부위염 관련 관절염 환자에 대한 이 약의 권장 투여 용량은 체중을 기반으로 한다.
|
체중 (kg) |
용량 |
|
15 kg 이상 30 kg 미만 |
20mg 격주 투여 |
|
30 kg 이상 |
40mg 격주 투여 |
3. 소아 판상 건선
소아 판상 건선에 대한 이 약의 권장 투여 용량은 체중을 기반으로 한다. 16주 이후에도 반응을 나타내지 않는 환자의 경우에는 투여 지속여부를 신중히 재고한다.
|
체중 (kg) |
용량 |
|
15 kg 이상 30 kg 미만 |
20mg을 처음 2회는 매주 피하주사하고, 이후에는 격주 투여 |
|
30 kg 이상 |
40mg을 처음 2회는 매주 피하주사하고, 이후에는 격주 투여 |
소아 판상 건선으로 4세 미만의 어린이에게 이 약을 사용한 적은 없다.
1. 경고
1) 감염 : 이 약의 투여 전, 투여 동안 및 투여 후 결핵을 포함한 감염에 대해 면밀히 모니터링해야 한다. 이 약의 배설에 5개월까지 소요될 수 있으므로 이 기간 동안 모니터링을 지속해야 한다. 만성 또는 국소화된 감염 등 활동성 감염 환자의 경우 감염이 조절될 때까지 이 약의 투여를 시작해서는 안 된다. 결핵에 노출된 환자 및 결핵의 위험이 높은 지역 또는 히스토플라스마증, 콕시디오이데스진균증, 또는 분아균증(blastomycosis) 같은 풍토병성 진균증의 위험이 높은 지역을 여행한 환자들은, 치료를 시작하기 전 이 약의 치료로 인한 위험 및 이익에 대하여 고려하여야 한다. 이 약의 투여 동안 새로운 감염이 발생한 환자는 면밀히 모니터링해야 하며, 완벽한 진단 평가를 거쳐야 한다. 만약 새로운 심각한 감염 또는 패혈증이 발생한 환자의 경우 감염이 조절될 때 까지 이 약의 투여를 중단해야 하며, 적절한 항균 또는 항진균 치료를 시작해야 한다. 재발성 감염 병력 또는 감염되기 쉬운 기저상태(면역억제제를 병용 투여 하는 경우 포함)에 있는 환자에게 이 약의 투여를 고려하는 의사는 주의를 기울여야 한다.
세균성, 진균성, 침습적 진균(파종성 또는 폐외 히스토플라스마증, 아스페르길루스증, 콕시디오이데스진균증), 바이러스성으로 인한 중대한 감염, 기생충에 의한 감염 또는 기타 기회감염이 TNF 저해제를 투여 받고 있는 환자들에게서 보고되었다. 또한 패혈증, 드물게 결핵, 칸디다증, 리스테리아증, 레지오넬라증 및 뉴모시스티스(pneumocystis)가 이 약을 포함한 TNF 저해제의 사용에서 보고되었다. 감염과 관련된 입원 또는 치명적인 결과가 보고되었으며, 심각한 감염은 대부분 선행 질환과 함께 감염을 일으키기 쉬운 면역억제제를 병용 투여한 환자에서 발생했다.
⓵ 심각한 감염
이 약을 투여받고 있는 환자에게 심각한 감염의 위험이 증가되는 경우가 임상시험에서 나타났으며, 시판 후 조사에서 이러한 사실을 뒷받침하는 보고가 있었다. 특히 폐렴, 신우신염, 패혈성 관절염, 패혈증과 같은 심각한 감염이 보고되었다.
⓶ 결핵
이 약을 투여받고 있는 환자에게서 결핵(재활성화 및 발병 포함)이 보고되었으며, 폐결핵 및 폐외결핵(예, 파종성)이 포함되었다.
이 약으로 치료를 시작하기 전 모든 환자의 활동성 또는 비활동성(잠복성) 결핵 감염에 대해 평가해야 한다. 이 평가에는 개인의 결핵 병력 또는 과거에 활동성 결핵 환자에 노출되었을 가능성과 과거 및 현재 면역억제요법에 대한 자세한 의학적 평가가 포함되어야 한다. 모든 환자에서 투베르쿨린 피부 검사와 흉부 X ‑선과 같은 적절한 스크리닝 시험을 실시해야 한다(국가별 권장사항을 적용할 수 있음).
특히 결핵이 널리 유행되는 지역에서 이민오거나 혹은 그곳을 여행한 환자들 또는 활동성 결핵 환자와 긴밀한 접촉이 있었던 환자들에서 진단되지 않은 잠복성 결핵이 가능하다는 것을 고려해야만 한다.
활동성 결핵으로 진단된 경우에는 이 약의 투여를 시작해서는 안 된다.
잠복성 결핵으로 진단된 경우에는, 이 약의 투여 시작 전 항결핵 예방요법과 함께 적절한 치료를 시작해야 하며, 국가별 권장사항에 따라야 한다. 결핵 테스트 결과가 음성일지라도 결핵의 중대한 위험인자를 갖고 있거나 여러 위험인자가 있는 환자 및 과거 활성 또는 잠복성 결핵의 병력이 있으나 적절한 치료가 확인되지 않은 환자 또한 이 약의 투여 시작 전에 항결핵 예방요법을 고려해야한다. 이 환자에 대한 항결핵 치료 시작은 잠복성 결핵 감염에 대한 위험과 항결핵 치료의 위험 두 가지를 모두 고려한 후에 결정되어야만 한다. 만약 필요하다면, 결핵치료의 경험이 있는 의사와 상담하여야만 한다.
잠복성 결핵 감염 환자의 항결핵 치료는 이 약을 투여받는 환자들의 결핵 재활성화 위험을 감소시킨다. 항결핵 예방요법에도 불구하고, 이 약을 투여 받는 동안 활동성 결핵이 발생한 경우가 있었다. 또한 잠복성 결핵 결과가 음성이었던 환자에서 이 약을 투여받고 활성 결핵이 발생하고, 활성 결핵 치료가 성공적으로 실시된 몇몇 환자에서도 TNF 저해제로 투여받는 동안 활성 결핵이 다시 나타난 바가 있다.
이 약으로 치료받는 환자는 잠복성 결핵이 위음성일 수 있으므로, 활성 결핵의 증상 및 징후가 모니터링되어야 한다. 투베르쿨린 위음성(false negative) 결과의 위험은 중증 질환자나 면역기능저하환자에서 특히 고려해야한다.
만약 이 약으로 치료하는 동안이나 치료 후 결핵을 암시하는 증상/징후(예. 지속성 기침, 쇠약/체중감소, 미열, 무기력)가 나타나는 경우 의사에게 즉시 알리도록 환자를 교육해야 한다.
⓷ 기타 기회감염
침습적 진균 감염을 포함한 기회감염이 이 약을 투여 받고 있는 환자들에서 관찰되었다. 이 감염들은 TNF 저해제를 투여 받고 있는 환자들에게서 일관되게 나타나지는 않았으며, 이는 적절한 치료의 지연을 야기하였고, 때로는 치명적인 결과를 초래하였다.
TNF 저해제를 투여받고 있는 환자들은 히스토플라스마증, 콕시디오이데스진균증, 또는 분아균증(blastomycosis), 아스페르길루스증, 칸디다증 및 기타 기회감염들에 좀 더 감수성이 높다. 발열, 권태감, 체중 감소, 발한, 기침, 호흡곤란 및/또는 폐 침윤, 또는 기타 심각한 (쇼크를 동반하거나 동반하지 않은) 전신질환이 나타난 환자는 즉시 진단 평가를 위한 의학적 조치를 취해야 한다.
진균증이 풍토병인 지역에 머물거나 여행한 환자의 경우, 전신적인 진균 감염의 징후가 나타나면, 침습적인 진균 감염을 의심해야 한다. 환자들은 히스토플라스마증 및 기타 침습적 진균 감염의 위험이 있으므로, 임상 의사들은 병원균이 확인되기 전까지 실험적 항진균 치료를 고려해야 한다. 활성 감염 상태인 몇몇의 환자들은 히스토플라스마증의 항원 및 항체 검사에서 음성을 나타낼 수 있다. 이러한 환자들에게 실험적 항진균 치료제의 투여는, 침습적 진균 감염의 치료 및 진단에 전문가인 의사와 상담하여 결정되어야 하며, 심각한 진균 감염의 위험 및 항진균 치료의 위험을 모두 고려하여야 한다. 심각한 진균 감염이 발생한 환자 또한 감염이 조절되기 전까지 TNF 저해제의 사용 중단이 고려되어야 한다.2) 악성종양과 림프증식성 질환
TNF 저해제에 대한 임상시험의 대조 시험동안 TNF 저해제를 투여받은 환자에서 대조군 환자에 비해 더 많은 림프종 사례가 관찰되었다. 그러나 발생이 드물었고 위약군 환자의 추적조사기간이 TNF 저해제를 투여받은 환자에 비해 더 짧았다. 더욱이 오래 지속된, 매우 활동성인, 염증성 질환을 가진 류마티스 관절염 환자는 림프종의 기저 위험(background risk)이 증가되어 있고 이는 위험 추정을 어렵게 한다. 이 약을 이용한 장기간 공개 임상시험에서 악성종양의 전체 발현율은 나이, 성별, 인종이 맞춰진 전체 인구에서 예상되는 발현율과 유사했다. 현재의 지식으로 TNF 저해제를 투여받는 환자에서 림프종 또는 다른 악성종양의 발생 위험 가능성을 배제할 수 없다.
TNF 저해제를 투여받는 어린이와 청소년들에서 악성종양이 보고되었고, 일부는 치명적이었다. 이 보고된 경우 중 대략 절반의 경우는 호지킨 및 비호지킨 림프종을 포함한 림프종이었다. 나머지 다른 경우들에서는 여러 다양한 악성 종양들이 나타났으며, 일반적으로 면역억제와 연관되어 드물게 나타나는 악성종양도 포함되었다. 악성종양은 중간값으로 치료 30개월 후에 나타났으며, 대부분의 환자가 면역억제제를 병용 투여하고 있었다. 이는 시판 후 조사에서 보고되었으며, 등록과 자발적인 시판 후 보고서를 포함한 여러 경로에서 나타났다.
이 약을 투여받은 환자에서, Hepatosplenic T ‑cell lymphoma(HSTCL)가 시판후 조사에서 매우 드물게 보고되었고, 공격적 림프종(종종 치명적임)이 드물게 확인되었다. 대부분의 환자들은 염증성대장질환의 치료를 위하여 아자티오프린 또는 6 ‑메르캅토푸린의 병용 투여는 물론이고 이전에 인플리시맵으로 치료를 받았었다. 이 약과 아자티오프린 또는 6 ‑메르캅토푸린의 병용 투여 시의 잠재적인 위험성을 주의깊게 고려해야 한다. 이 약 복용환자에서 HSTCL의 발생 위험 가능성은 배제할 수 없다
악성종양의 병력이 있는 환자를 포함하거나 이 약 투여 중 악성종양이 발생한 환자에게 투여를 지속한 시험은 실시되지 않았다. 따라서 이러한 환자군에 투여를 고려시 추가적인 주의를 기울여야 한다. 모든 환자, 특히 과다한 면역억제제 치료의 병력이 있는 환자나, PUVA 치료의 병력이 있는 건선 환자들은 이 약의 투여 시작 전 및 투여 기간 동안 비흑색종 피부암 존재 여부에 대한 검사를 받아야 한다.
TNF 저해제를 류마티스 관절염과 다른 적응증에서 사용하여 급성 및 만성 백혈병이 나타난 경우가 보고되었다. 류마티스 관절염이 있는 환자들은 TNF 저해제를 투여받지 않더라도 다른 사람들에 비해 백혈병이 발병될 확률이 (2배까지) 높을 수 있다.
중등증에서 중증의 만성 폐쇄성 폐질환(COPD) 환자들을 대상으로 다른 TNF 저해제인 인플릭시맵을 사용하여 실시한 탐색적 임상시험에서, 현재 흡연중이거나 기존에 흡연하였던 사람의 경우 비교군의 환자보다 인플릭시맵 치료를 받은 환자군에서 악성 종양이 많이 보고되었다. 과도한 흡연으로 인해 악성종양의 위험성이 증가한 환자는 물론 만성 폐쇄성 폐질환(COPD) 환자에 대한 TNF ‑α 저해제 치료 고려 시 주의가 필요하다.
이 약이 형성이상증이나 대장암의 발생 위험에 영향을 미치는지 여부는 현재 알려지지 않았다. 형성이상증 또는 대장암종의 위험이 증가되었거나(예, 오래 지속되는 궤양성 대장염 또는 원발성 경화성 담관염), 형성이상증 또는 대장암종의 병력이 있는 모든 궤양성 대장염 환자들은 치료 시작 전 및 치료 중에 주기적으로 형성이상증에 대해 확인해야한다. 대장 내시경검사 및 생검 등 국가별 권장사항에 따라 평가한다.
3) B형 간염 재활성화
이 약을 포함한 TNF 저해제의 사용으로 B형 간염 만성 보균자에서 병증이 재활성화 될 수 있으며, 일부는 치명적이었다. 이 보고내용의 대부분은 다른 면역억제제를 함께 투여받는 환자에게서 나타났으며, 이는 또한 B형 간염 재활성화를 일으킬 수 있다. B형 간염의 감염 위험이 있는 환자는 이 약을 투여 받기 전 감염 여부에 대한 검사를 하여야 한다. B형 간염 보균자로 확인된 환자에게 TNF 저해제를 처방하는 경우, 의사는 주의를 기울여야 한다. B형 간염 보균자이고, TNF 저해제의 치료를 요하는 환자는 이 약을 투여 받는 동안과 투여가 끝난 후 수 개월 동안 감염의 증상/징후를 철저히 관찰해야 한다. B형 간염 보균자의 재활성화를 방지하기 위해 항바이러스제와 TNF 저해제를 함께 사용하는 것에 대한 적절한 치료법은 알려진 바 없다. B형 간염이 재활성화 된 경우 이 약의 투여를 중지하고 적절한 치료와 함께 항바이러스제 치료를 시작해야 한다.
4) 신경학적 반응
이 약을 포함한 다른 TNF 저해제는 드물게, 다발성 경화증 및 시신경염을 포함한 중추신경계 탈수초성질환(demyelinating disease)과 길랑 바레 증후군(Guillian Barre syndrome)을 포함한 말초 탈수초성질환의 임상 증상 및/또는 방사선 증거의 새로운 발현 혹은 악화와 관련이 있다. 중추신경계 및 말초신경계 탈수초성질환의 기왕력이 있거나 최근에 발현된 환자에게 이 약의 투여를 고려하는 경우 주의를 기울여야 한다. 이러한 이상이 진행되면 이 약의 중단을 고려하여야 한다.
중등도 포도막염과 중추 신경계 탈수초성장애 간의 연관성은 알려져있다. 비 ‑감염성 중등도 포도막염 환자에서 이 약 치료 전 중추신경계 탈수초성 장애 여부를 측정하는 신경학적 평가를 하여야 한다.
2. 다음 환자에는 투여하지 말 것
1) 이 약 또는 이 약의 성분에 과민증인 환자
2) 활동성 결핵 또는 패혈증, 기회감염과 같은 다른 중증 감염이 있는 환자(경고항 참조)
3) 중등도 내지 중증의 심부전(NYHA class III/IV) 환자
3. 이상반응
1) 이 약은 치료적 확증 대조 및 공개 임상시험을 통해 60개월 이상에 걸쳐 9,506명의 환자에 대하여 연구되었다. 이 임상시험들에는 단기 및 장기 질병상태를 보이는 류마티스 관절염 환자뿐만 아니라 건선성 관절염 및 축성 척추관절염(강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염) 환자 및 크론병 환자, 궤양성 대장염 환자 및 건선 환자, 화농성 한선염 환자, 포도막염 환자와 소아 특발성 관절염 (다관절 소아 특발성 관절염 및 골부착부위염 관련 관절염) 환자가 포함되었다. 표 1의 자료는 대조 임상시험 기간 동안 이 약을 투여받은 6,089명의 환자와 위약 또는 활성 대조약을 투여받은 3,801명의 환자를 포함한 주요 대조 임상시험 결과를 근거로 한 것이다. 주요 임상시험 중 이중 눈가림, 대조 임상시험 기간 동안 이상사례로 인해 투여를 중단한 환자의 비율은 이 약 투여군이 5.9%이고 대조군이 5.4%였다.
일반적으로 소아환자에서의 이상사례는 성인 환자에서 나타난 이상사례의 빈도 및 타입과 유사하였다.
이 약을 이용한 대조 임상시험에서 가장 빈번히 나타났던 이상사례들 중 하나를 근거로 약 13%의 환자들이 주사부위반응을 경험할 것으로 예상할 수 있다.
아래 표 1은 주요 임상시험에서 임상적 및 실험실 검사 모두 적어도 이 약과 인과관계의 가능성이 있는 약물이상반응을 발현부위와 빈도별(매우 자주 ‑1/10 이상 ; 자주 ‑1/100 이상 및 1/10 미만 ; 때때로 ‑1/1,000 이상 및 1/100 미만 ; 드물게 ‑1/1,000 미만)로 요약하였다. 각각의 발현빈도 그룹 안에, 약물이상반응은 심각성이 감소하는 순서로 배열되었다. 이 약의 여러 적응증에서 가장 자주 나타난 약물이상반응들이 포함되었다. 신체기관 열에서 (*)로 표시된 부분은 1. 경고, 4. 일반적 주의 항에서 자세한 내용을 확인할 수 있다.
2) 베체트 장염 : 일본의 베체트 장염 연구에서의 안전성 프로파일은 이미 연구된 이 약의 안전성 프로파일과 유사하였다.
3) 화농성 한선염: 이 약으로 매주 치료받은 화농성 한선염 환자의 안전성 프로파일은 이미 알려진 이 약의 안전성 프로파일과 유사하였다.
4) 포도막염: 이 약으로 치료받은 비 ‑감염성 포도막염 환자의 안정성 프로파일은 이미 알려진 이 약의 안전성 프로파일과 유사하였다.
<표 1. 임상시험 중 약물이상반응> <! ‑ ‑표 ‑ ‑><! ‑ ‑표 ‑ ‑><! ‑ ‑표 ‑ ‑>
◎ 기타 이상사례
이 약으로 치료를 받은 류마티스 관절염 환자에게 5% 미만의 드문 발생률로 나타난 기타 중대한 이상사례는 다음과 같다.
전신 : 발열, 감염, 극심한 통증, 골반 통증, 패혈증, 수술, 가슴통증, 결핵의 재발
심혈관계 : 부정맥, 심방 세동, 심혈관 장애, 흉통, 울혈성 심부전, 관상동맥 이상, 심정지, 고혈압성 뇌병증, 심근경색증, 심계항진, 심낭삼출, 심낭염, 실신, 빈맥, 혈관 장애
콜라겐 질환 : 홍반성 루푸스 증후군
소화기계 : 담낭염, 담석증, 식도염, 위염, 위장장애, 위장출혈, 간 괴사, 구토
내분비계 : 부갑상선 장애
혈액계 : 무과립구증, 과립구감소증, 백혈구감소증, 림프종 유사반응, 범혈구감소증, 적혈구증가증
대사 및 영양장애 : 탈수증, 치유 이상, 케톤증, 파라프로테인혈증, 말초부종
근골격계 : 관절염, 뼈장애, 뼈골절(자연적인 경우가 아닌), 골괴사, 관절장애, 근육경련, 중증근무력증, 화농성 관절염, 윤활막염, 힘줄 장애
종양 : 샘종, 유방 ◦ 위장관 ◦ 피부 ◦ 비뇨생식과 같은 암종 및 기타(림프종, 흑색종)
신경계 : 착란, 다발성 경화증, 감각이상, 경막하혈종, 떨림
호흡기계 : 천식, 기관지연축, 호흡곤란, 폐 장애, 폐기능 감소, 흉막 삼출, 폐렴
피부 및 부속물 : 연조직염, 얕은 연조직염, 대상포진
특수감각 : 백내장
혈전증 : 다리혈전
비뇨생식기계 : 방광염, 신장 결석, 월경 장애, 신우신염
5) 주사부위반응 : 성인 및 소아에 대한 주요 대조 임상시험에서 이 약을 투여한 환자의 12.9%가 이 약을 투여했을 때 가장 흔히 나타난 이상사례인 주사부위반응(홍반 및/또는 가려움증, 출혈, 통증 또는 부종)을 나타낸 반면, 위약 또는 활성 대조군을 투여한 환자에서는 7.2%에서 이러한 반응이 나타났다. 주사부위반응은 일반적으로 투여 중단이 필요하지는 않았다.
6) 감염 : 성인 및 소아에 대한 주요 대조 임상시험에서, 감염률은 이 약 투여군이 1.51/환자 ‑년(patient year)이고 위약 및 활성 대조약 투여군은 1.46/환자 ‑년이었다. 감염은 주로 비인두염, 상기도 감염 및 부비동염으로 구성되었다. 대부분의 환자들은 감염이 소실된 후 이 약을 계속 투여하였다. 심각한 감염 발생률은 이 약 투여군이 0.04/환자 ‑년이고 위약 및 활성 대조약 투여군은 0.03/환자 ‑년이었다. 이 약을 이용한 성인 및 소아에 대한 대조 및 공개 임상시험에서 심각한 감염(드물게 발생한 치명적 감염을 포함)이 보고되었으며, 이는 결핵(속립성 및 폐 이외 부위 포함) 및 침습적 기회 감염(예. 파종성 히스토플라스마증, 폐포자충 폐렴, 아스페르길루스증 및 리스테리아증)을 포함한다. 결핵 사례의 대부분은 치료 시작 후 최초 8개월 이내에 발생하였으며, 이는 잠복 질환의 재발을 반영하는 듯하다.
7) 악성종양과 림프증식성 질환 : 소아 특발성 관절염(다관절형 소아 특발성 관절염 및 골부착부위염 관련 관절염) 환자를 대상으로 655.6 환자 ‑년 노출된 249명에서의 임상시험 동안 악성종양이 관찰되지 않았다. 또한, 소아 크론병 환자를 대상으로 498.1환자 ‑년 노출된 192명에서의 임상시험 동안 악성종양이 관찰되지 않았다.
판상 건선 소아 환자를 대상으로 80.0 환자 ‑년 노출된 77명의 소아 환자에서의 임상시험 동안 악성 종양이 관찰되지 않았다. 중등도에서 중증의 류마티스 관절염, 건선성 관절염, 축성 척추관절염(강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염), 크론병, 궤양성대장염, 화농성 한선염, 건선, 포도막염 환자를 대상으로 12주 이상 실시한 성인에 대한 주요 대조 임상시험에서 림프종, 비흑색종 피부암 이외에도 악성종양이 투여군 5,291명 중에서 6.8/1,000 환자 ‑년, 대조군 3,444명 중에서 6.3/1,000 환자 ‑년의 비율로 생성되었다(치료기간의 중간값은 이 약의 투여군에서는 4.0개월, 대조군에서는 3.8개월이었다). 비흑색종 피부암은 투여군에서 8.8/1,000 환자 ‑년 및 대조군에서 3.2/1,000 환자 ‑년의 비율로 생성되었다. 피부암중에서 편평세포암종은 투여군에서 2.7/1,000 환자 ‑년, 대조군에서 0.6/1,000 환자 ‑년의 비율로 생성되었다. 림프종의 비율은 투여군에서 0.7/1,000 환자 ‑년, 대조군에서 0.6/1,000 환자 ‑년이었다.
대조 임상시험과 진행 중 또는 완료된 공개 연장 시험에서, 림프종 및 비흑색종 피부암 이외에 관찰된 악성종양의 발생률은 약 8.5/1,000 환자 ‑년이다. 비흑색종 피부암의 관찰된 비율은 약 9.6/1,000 환자 ‑년이며, 림프종이 관찰된 비율은 약 1.3/1,000 환자 ‑년이다. 이 임상시험기간의 중앙값은 약 3.3년이며, 최소 1년 동안 이 약을 투여받은 6,427명의 환자들 또는 26,439.6 환자 ‑년 투여를 초과하는 치료 시작 1년 이내에 악성종양으로 발전한 환자들을 포함했다.
2003년 1월부터 2010년 12월까지의 시판 후 경험 중 주로 류마티스 관절염 환자에서 보고된 악성종양의 발생 비율은 약 2.7/1,000 환자 ‑년 이었다. 비흑색종 피부암과 림프종의 발생 비율은 각각 약 0.2 및 0.3/1,000 환자 ‑년이었다.
8) 자가항체 : 류마티스 관절염에 대한 5건의 임상시험 중 여러 시점에서 혈청 내 자가항체에 대한 검사를 실시했다. 이러한 적절히 디자인된 대조 임상시험에서 기저 항 핵항체(ANA, anti ‑nuclear antibody)가 음성이었던 환자 중 이 약 투여군의 11.9%와 위약 및 활성 대조군의 8.1%에서 24주째 양성을 나타냈다. 모든 류마티스 관절염과 건선성 관절염에 대해 이 약을 투여받은 3,441명 중 2명에서 새롭게 발생된 루푸스양 증후군임을 시사하는 임상 증상이 나타났으며 이 환자는 투여 중단 후 개선되었다. 루푸스성 신장염이나 중추신경계 증상이 나타난 환자는 없었다.
9) 건선 : 발현 및 악화
이 약을 포함하여 TNF 저해제를 사용 시, 농포성 건선 및 손발바닥 건선을 포함하여 건선이 첫 발현되는 경우와 이미 존재하던 건선이 악화되는 경우들이 보고되었다. 이 환자들의 대다수는 면역억제제(예. MTX, 코르티코스테로이드)를 병용하고 있었으며, 일부는 입원치료가 필요했다. 대부분의 환자는 TNF 저해제의 복용을 중단함으로써 건선이 호전되었다. 일부 환자들은 이후, 다른 TNF 저해제를 재 투여했을 때 건선이 재발하였다. 국소 치료에도 불구하고 증상이 호전되지 않거나, 악화되는 심각한 경우에는 이 약의 투여 중지를 고려해야만 한다.
10) 간 효소 상승
◦ 류마티스 관절염 임상시험 : 류마티스 관절염에 대한 대조 임상시험에서(4건의 임상시험), ALT의 상승은 이 약 또는 위약을 투여한 환자에서 유사했다. 초기 류마티스 관절염 환자에서(3년 미만의 유병기간)(1건의 임상시험), ALT의 상승은 병용 투여군(이 약/메토트렉세이트)에서 메토트렉세이트 단독군 또는 이 약 단독군에 비해 더 자주 발생했다. 다관절형 소아 특발성 관절염 환자를 대상으로 한 임상시험에서 일부 트랜스아미나제의 상승이 나타났으나 위약과 이 약 투여군에서 같거나 유사했으며, 대부분은 메토트렉세이트와 병용 투여 시 나타났다.
◦ 건선성 관절염 임상시험 : ALT의 상승은 류마티스 관절염 임상시험의 환자에 비해 건선성 관절염 환자에 대한 임상시험(2건의 임상시험)에서 더 자주 발생하였다.
◦ 모든 류마티스 관절염, 다관절형 소아 특발성 관절염 및 건선성 관절염 임상시험에서, 상승된 ALT를 나타내는 환자들은 증상이 없었고, 대부분 상승이 일시적이며 투여 지속시 소실되었다.
◦ 크론병 및 궤양성 대장염 임상시험 : 대조 임상시험에서 이 약 투여군과 대조군간의 ALT상승은 유사하였다.
체중에 따라 조정된 유도 요법 후 체중에 따라 조정된 유지 요법(최대 52주)의 유효성과 안전성을 평가한 소아 크론병의 3상 임상시험에서, 시험대상자의 2.6%(5/192)에서 ≧ 3 X ULN의 ALT 상승이 발생하였고, 이 중 4명은 베이스라인에서 면역억제제를 병용 투여하였다.
◦ 건선 임상시험 : 건선에 대한 대조 임상시험에서, 투여군과 대조군간의 ALT 상승은 유사하였다.
◦ 화농성 한선염 임상시험 : 화농선 한선염에 대한 대조 임상시험에서, 투여군의 0.3%와 대조군의 0.6%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다.
◦ 소아 특발성 관절염 임상시험 : 4 ‑ 17세 다관절형 소아 특발성 관절염 환자와 6 ‑ 17세 골부착부위염 관절염 환자의 3상 대조 임상 시험에서, 투여군의 6.1%와 대조군의 1.3%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다. 대부분의 ALT 상승은 메토트렉세이트 병용 사용에 따라 발생하였다. 2세에서 4세 미만의 다관절형 소아 특발성 관절염 환자에 대한 3상 임상시험에서, ≧ 3 X ULN의 ALT 상승은 발생하지 않았다.
◦ 판상 건선 임상시험 : 판상 건선 소아 환자에 대한 3상 임상시험에서 ≧ 3 X ULN의 ALT 상승은 발생하지 않았다.
◦ 포도막염 임상시험: 포도막염에 대한 대조 임상시험에서, 투여군의 2.4%와 대조군의 2.4%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다.
모든 적응증의 임상시험에서 상승된 ALT를 나타내는 환자들은 증상이 없었고, 대부분 상승이 일시적이며 투여 지속 시 소실되었다. 그러나 간부전을 포함한 중증의 간손상이 이 약을 포함한 TNF 저해제를 투여받은 환자의 시판 후 보고에서 아주 드물게 나타났다. 이 약 투여와의 관계는 명백하지 않다.
11) 아자티오프린 또는 6 ‑메르캅토푸린과의 병용 투여 : 성인 크론병 임상시험에서, 아자티오프린/6 ‑메르캅토푸린과 병용 투여 시 이 약의 단독 투여에 비해 악성 종양 및 심각한 감염과 관련된 이상사례의 발생률이 더 높게 나타났다.
12) 시판 후 조사 또는 4상 시험에서 보고된 추가 이상사례 : 4상 시험과 시판 후 조사에서 보고된 추가 이상사례는 다음 표 2와 같다.
<표 2. 시판 후 조사 및 제4상 시험에서의 이상사례>
13) 재심사에 따른 국내 시판 후 조사결과
⓵ 국내에서 6년 동안 류마티스 관절염, 건선성 관절염, 강직성 척추염, 크론병, 건선환자를 대상으로 실시한 시판 후 조사
국내에서 1,698명(류마티스 관절염 652명, 건선성 관절염 47명, 강직성 척추염 925명, 크론병 73명, 건선 1명)을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 10.07%(171명/1,698명, 238건)이었고, 이 중 이 약과 인과관계를 배제할 수 없는 약물이상반응 발현율은 6.83%(116명/1,698명, 141건)이며, 주사부위 반응 19건(1.11%), 가려움(증) 9건(0.53%), 발진 8건(0.47%), 대상포진, 상기도 감염, 두드러기 각 4건(0.24%), 신우신염, 홍반, 복통, 어지러움, 두통 각 3건(0.18%), 연조직염, 폐결핵, 결핵성 복막염, 폐렴, 결핵, 피부반응, 알레르기성 피부염, 림프종, 근육통, 기침, 림프절병증 각 2건(0.12%), 모낭염, 신장결핵, 비염, 편도염, 말라리아, 기관지 폐렴, B형 간염, 코 인두염, 감염성 윤활낭염, 진균성 감염, 크립토코쿠스 폐렴, 농포성 발진, 가려움 발진, 피부염, 탈모(증), 피부병변, 무력(증), 구토, 설사, 복막염, 구역, 고창, 갑상선암, 기관지 신생물, 급성 골수성 백혈병, 골관절염, 관절종창, 폐출혈, 콧물, 간손상, 간독성, 간기능 이상, 찔림 알레르기, 현기증, 월경 장애, 포도막염 각 1건(0.06%)이 보고되었다.
중대한 이상사례 발현율은 2.77%(47명/1,698명, 63건)이며, 신우신염, 복통이 각 4건이었으며 폐렴, 폐 결핵, 대상포진, 결핵성 복막염, 결핵, 설사, 구토, 가려움(증)이 각각 2건이며 그 외 신장결핵, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 항문 농양, 복막 농양, 결장염, 장관 천공, 복막염, 장관 경색, 장피부누공, 구역, 홍반, 발진, 대뇌출혈, 마비, 치매, 골관절염, 강직성 척추염, 등 통증, 갑상선암, 급성 골수성 백혈병, 직장암, 무력(증), 발열, 통증, 추골 탈구, 골절, 폐 출혈, 호흡곤란, 무릎 수술, 소장 절제(술), 혈중 크레아티닌 증가, 혈색소 감소, 간독성, 찔림 알레르기, 심근경색증, 빈혈, 탈수는 각 1건이 보고되었다.
이 중 이 약과 인과관계를 배제할 수 없는 중대한 약물이상반응 발현율은 1.47%(25명/1,698명, 30건)이며, 신우신염 3건, 복통, 폐결핵, 대상포진, 결핵성 복막염, 결핵, 가려움(증) 각 2건, 폐렴, 신장결핵, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 복막염, 홍반, 발진, 골관절염, 갑상선암, 급성 골수성 백혈병, 폐출혈, 혈중 크레아티닌 증가, 간독성, 찔림 알레르기 각 1건이 보고되었다.
예상하지 못한 이상사례 발현율은 3.24%(55명/1,698명, 61건)이며, 설사, 콧물 각 5건, 관절 종창, 포도막염 각 3건, 등 통증, 사지 통증, 감각 이상, 무력(증), 림프절병증 각 2건, 결장염, 복막염, 장관 경색, 위장관 궤양, 장피부누공, 명치불쾌감, 치루, 고창, 모낭염, 비염, 편도염, 말라리아, 기관지 폐렴, 감염성 윤활낭염, 크립토코쿠스 폐렴, 농포성 발진, 항문 농양, 복막 농양, 경부 통증, 강직성 척추염, 대뇌출혈, 마비, 치매, 신경근병증, 기억장애, 폐 출혈, 눈마름, 압통, 종양 절제(술), 간독성, 추골 탈구, 현기증, 피부병변은 각각 1건이 보고되었다.
이 중 이 약과 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 1.24%(21명/1,698명, 21건)이며, 림프절병증은 2건, 설사, 복막염, 고창, 모낭염, 비염, 편도염, 말라리아, 기관지 폐렴, 감염성 윤활낭염, 크립토코쿠스 폐렴, 농포성 발진, 관절 종창, 콧물, 폐 출혈, 포도막염, 무력(증), 간독성, 현기증, 피부병변은 각각 1건이 보고되었다. 중대하고 예상하지 못한 이상사례 발현율은 1.12%(19명/1,698명, 20건)이며, 설사는 2건, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 항문 농양, 복막 농양, 결장염, 복막염, 장관 경색, 장피부누공, 대뇌출혈, 마비, 치매, 강직성 척추염, 등 통증, 무력(증), 추골 탈구, 폐 출혈, 간독성은 각 1건이 보고되었고, 중대하고 예상하지 못한 약물이상반응 발현율은 0.35%(6명/1,698명, 6건)이며, 복막염, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 폐 출혈, 간독성 각 1건이 보고되었다.
⓶ 국내에서 4년 동안 소아 특발성 관절염(다관절형 소아 특발성 관절염, 골부착부위염 관련 관절염) 환자 28명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 21.43%(6명/28명, 8건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 17.86% (5명/28명, 7건) 이며, 독감 10.71% (3명/28명, 3건) 등통증, 관절부기, 두드러기, 발열 각 3.57% (1명/28명, 1건)이 보고되었다. 중대한 이상사례 발현율은 3.57%(1명/28명, 1건)이며, 홍반이 3.57% (1명/28명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응은 확인되지 않았다.
예상하지 못한 이상사례 발현율은 3.57% (1명/28명, 1건)이었고, 이 1건은 관절부기였으며 이는 본 제와 인과관계를 배제할 수 없다. 중대하고 예상하지 못한 이상사례 및 중대하고 예상하지 못한 약물이상반응은 확인되지 않았다.
⓷ 국내에서 4년 동안 소아 크론병 환자 143명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 18.18%(26명/143명, 47건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 14.69%(21명/143명, 26건)이며, 백혈구감소증 2.80%(4명/143명, 4건), 발진 2.10%(3명/143명, 3건), 설사, ALT증가, AST증가 각 1.40%(2명/143명, 2건), 대변내혈액, 마비성장폐쇄, 장천공, 가려움증, 국소피부반응, 두드러기, 상세불명의간기능검사이상, 충수돌기염, 권태, 주사부위홍반, 칸디다증, 신우신염, 모낭염이 각 0.70%(1명/143명, 1건) 보고되었다.
중대한 이상사례 발현율은 5.59%(8명/143명, 13건)이며, 복통 1.40%(2명/143명, 2건), 창자괴사, 대변내혈액, 마비성장폐쇄, 장폐쇄, 장천공, 소장폐쇄, 충수돌기염, 복막염, 열, 칸디다증, 신우신염이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응 발현율은 3.50%(5명/143명, 5건)이며, 마비성장폐쇄, 장천공, 충수돌기염, 칸디다증, 신우신염이 각 0.70%(1명/143명, 1건) 보고되었다.
예상하지 못한 이상사례 발현율은 6.29%(9명/143명, 17건)이며, AST증가 2.10%(3명/143명, 3건), 창자협착증 1.40%(2명/143명, 2건), 대장혈종, 위장염, 창자괴사, 마비성장폐쇄, 장폐쇄, 소장폐쇄, 충수돌기염, 헬리코박터파이로리위염, C반응단백질증가, 적혈구침강속도증가, 난치성통증, 명시안된수술후합병증이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 2.80%(4명/143명, 4건)이며, AST증가 1.40%(2명/143명, 2건), 마비성장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다.
중대하고 예상하지 못한 이상사례 발현율은 3.50%(5명/143명, 5건)이며, 창자괴사, 마비성장폐쇄, 장폐쇄, 소장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대하고 예상하지 못한 약물이상반응 발현율은 1.40%(2명/143명, 2건), 마비성장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다.
⓸ 국내에서 4년 동안 성인 화농성 한선염 환자 및 소아 판상 건선 환자 19명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 42.11%(8명/19명, 12건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 31.58%(6명/19명, 10건)이며, 한선염 15.79%(3명/19명, 4건), 여드름 5.26%(1명/19명, 2건), 소양증, 종기, 편도염, 발열이 각각 5.26%(1명/19명, 1건)으로 보고되었다. 중대한 이상사례 및 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응은 확인되지 않았다. 예상하지 못한 이상사례 발현율은 21.05%(4명/19명, 6건)이었고, 한선염 15.79%(3명/19명, 4건), 여드름 5.26%(1명/19명, 2건)으로 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 21.05%(4명/19명, 6건)이었다. 중대하고 예상하지 못한 이상사례 및 중대하고 예상하지 못한 약물이상반응은 확인되지 않았다.
⓹ 국내에서 포도막염에 대해 재심사를 위하여 4년 동안 155명을 대상으로 실시한 시판 후 조사 결과, 이상사례의 발현율은 인과관계와 상관없이 8.39%(13명/155명, 총 25건)로 보고되었다. 이 중 이 약과 인과관계를 배제할 수 없는 약물이상반응 발현율은 3.23%(5명/155명, 6건)이며, 안구 불편감, 주사부위 과민증, 주사 부위 통증, 지각 이상, 근육통, 습진 각 0.65%(1명/155명, 1건) 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응은 없었고, 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 1.29%(2명/155명, 2건) 안구 불편감, 주사부위 과민증 각 0.65%(1명/155명, 1건) 보고되었다.
➅ 이 약에 대한 국내 재심사 이상사례 및 자발적 부작용 보고자료를 국내 시판 허가된 모든 의약품을 대상으로 보고된 이상사례 보고자료(1989 ‑2021.5.31)와 재심사 종료시점에서 통합평가한 결과, 다른 모든 의약품에서 보고된 이상사례에 비해 이 약에서 통계적으로 유의하게 많이 보고된 이상사례 중 새로 확인된 것들은 다음과 같다. 다만, 이 결과가 해당성분과 다음의 이상사례 간에 인과관계를 입증된 것을 의미하는 것은 아니다.
◦ 근육 ‑골격계 이상 : 관절통, 관절병증
◦ 대사 및 영양계 이상 : 체중증가, 고지혈증
◦ 전신 및 투여부위 이상 : C반응단백질증가, 주사부위 덩어리, 주사부위 멍듦, 상태악화
◦ 호흡기계 이상 : 가래증가
◦ 정신계 이상 : 식욕증가
◦ 중추 및 말초신경계 이상 : 보행이상
◦ 생식기계 이상(여성) : 월경과다
◦ 소화기계 이상 : 흑색변
◦ 감염 : 농양, 종기증
◦ 피부 및 피하조직계 이상 : 각화과다증
14) 국내 관찰연구 결과
국내에서 베체트장염에 대해 6년 동안 50명을 대상으로 관찰 연구한 결과, 이상사례의 발현율은 인과관계와 상관없이 72%(36/50, 총 122건)로 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응 및 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 발현 빈도에 따라 아래 표에 나열하였다.
4. 일반적 주의
1) 알레르기 반응 : 임상시험 동안 이 약의 피하주사로 인한 심각한 알레르기 반응은 드물게 보고되었다. 이 약 투여 후 아나필락시스를 포함한 중대한 알레르기 반응이 보고된 바 있다. 만일 아나필락시스 반응이나 다른 심각한 알레르기 반응이 나타나면 이 약의 투여를 즉시 중단하고 적절한 치료를 시작해야 한다.
2) 면역억제 : 이 약을 투여받은 64명의 류마티스 관절염 환자에 대한 시험에서 지연성 과민반응의 억제, 면역글로불린 억제 또는 effector T ‑와 B세포, NK ‑세포, 단핵세포/대식세포 및 호중구의 수적 변화의 증거는 없었다.
3) 혈액학적 반응 : TNF 저해제에서 드물게 재생불량성빈혈을 포함한 범혈구감소증이 보고되었다. 의학적으로 중요한 혈구감소증(예. 저혈소판혈증, 백혈구감소증)을 포함한 혈액계의 이상사례가 이 약에서 보고되었다. 이러한 보고의 이 약에 대한 원인 상관관계는 불분명하다. 이 약을 투여하는 동안 혈액질환을 암시하는 징후 또는 증상(예, 지속적인 발열, 타박상, 출혈, 창백)이 발현되는 경우 즉각적인 의학적 처치를 받도록 모든 환자를 교육해야 한다. 심각한 혈액학적 이상이 확정된 환자는 이 약의 투여 중단을 고려해야 한다.
4) 예방접종 : 이 약 또는 위약을 투여한 226명의 성인 류마티스 관절염 환자에 대한 시험에서 표준 23가 폐구균 백신과 3가 인플루엔자 백신에 대한 항체반응과 유사한 반응이 관찰되었다. 이 약을 투여받은 환자에게 생백신을 투여한 경우 이에 의한 이차적인 감염 전파에 대한 이용 가능한 자료는 없다. 이 약을 투여 받고 있는 환자에게 생백신을 제외한 다른 백신을 병용 투여할 수 있다.
5) 울혈성 심부전 : 다른 TNF 저해제를 사용한 임상시험에서 울혈성 심부전의 악화와 울혈성 심부전으로 인한 사망률 증가가 관찰되었다. 이 약을 투여받은 환자에서도 울혈성 심부전이 악화된 사례가 보고되었다. 경증 심부전(NYHA classI/II) 환자에게 이 약 투여시 주의해야 하며 중등도 또는 중증 심부전 환자에게 투여해서는 안 된다. 울혈성 심부전이 새롭게 발생하거나 증상이 악화된 환자의 경우 이 약의 투여를 중단해야 한다.
6) 자가면역 과정 : 이 약의 투여로 자가면역 항체가 형성될 수 있다. 이 약의 장기투여가 자가면역질환 발생에 미치는 영향은 알려지지 않았다. 만일 이 약으로 치료 후 환자에게서 루푸스양 징후를 암시하는 증상을 나타내고 dsDNA에 대한 항체가 양성이라면 이 약의 투여를 중지해야 한다.
7) 생물학적 DMARDs 또는 TNF 저해제와의 병용투여 : 에타너셉트(다른 TNF 저해제)와 아나킨라를 병용 투여한 임상 시험에서 심각한 감염이 관찰되었으며, 에타너셉트 단독 요법에 비해 추가적인 유익성이 없었다. 에타너셉트와 아나킨라의 병용 시 관찰된 이상사례의 특성 때문에, 아나킨라와 다른 TNF 저해제와의 병용으로 인해 유사한 독성이 있을 수 있다. 이 약과 아나킨라의 병용 투여가 권장되지 않는다. 감염 및 기타 잠재적 약물학적 상호작용의 위험 증가 가능성으로 이 약과 다른 생물학적 DMARDs (예. 아나킨라 및 아바타셉트) 또는 다른 TNF 저해제와의 병용투여는 권장되지 않는다.
8) 수술 : 이 약을 투여받은 환자에서 수술 절차의 안전성 경험은 제한적이다. 만일 수술 절차가 계획된다면 이 약의 긴 반감기를 고려해야 한다. 이 약 투여동안 수술이 필요한 환자는 감염에 대해 면밀히 모니터링하고 적절한 조치를 취해야 한다. 이 약 투여 동안 관절성형술을 받는 환자에 대한 안전성 경험은 제한적이다.
9) 소장 폐색증 : 크론병의 치료에 반응하지 않은 환자는 외과적 치료를 요하는 고정된 섬유성 협착을 보일 수 있다. 이 약이 협착을 일으키거나 악화시키지 않는다는 것을 나타내는 자료가 있다.
10) 간 ◦신장애 환자 : 이러한 환자군에 대해서 연구되지 않았으며 권장 용량이 없다.
11) 이 약이 운전이나 기계 조작 능력에 미치는 영향에 대해서는 연구된 바 없다.
5. 상호작용
1) 이 약은 류마티스 관절염 환자에서 단독요법 및 메토트렉세이트와의 병용 요법 모두에 대해 연구되었다. 시험결과 이 약 또는 메토트렉세이트의 용량조절 필요성은 나타나지 않았다. 공식적인 약동학 시험에서 이 약과 메토트렉세이트 이외의 약물과의 상호작용을 평가하지 않았다. 임상시험에서 이 약과 일반적으로 사용되는 DMARDs(sulfasalazine, hydrochloroquine, leflunomide 및 parenteral gold), 글루코코르티코이드, 살리실산염, 비스테로이드성항염증제 또는 진통제를 병용 투여 시 상호작용이 나타나지 않았다.
이 약 단독투여에 비해 메토트렉세이트와 병용 투여 시 항체 생성률이 더 낮았다. 메토트렉세이트를 투여하지 않고 이 약 단독투여시 항체 생성률과 이 약의 제거율이 증가했으며, 이 약의 효과는 감소했다.
2) 생백신과 이 약의 병용 투여는 권장되지 않는다.
3) 아나킨라와 이 약의 병용 투여는 권장되지 않는다.
4) 약물/실험실 검사 상호작용 : 이 약과 실험실 검사 사이에 알려진 간섭은 없다.
6. 임부 및 수유부에 대한 투여
1) 원숭이에 대한 발생독성시험에서 모체 독성, 배아 독성 또는 최기형성이 나타나지 않았다. 출생후 독성과 수태능에 미치는 영향에 대한 비임상시험 자료는 없다. TNF 알파 저해 작용으로 인해 임신기간 중 이 약 투여는 신생아의 정상 면역반응에 영향을 줄 수 있다. 임신기간 동안 이 약은 태아에 대한 잠재적 유익성이 잠재적 위험성을 상회하는 경우에만 임부에게 투여해야한다. 임신할 가능성이 있는 여성은 임신을 예방하기 위한 적절한 피임요법 사용과 이 약의 최종 투여 후 최소 5개월간 피임을 지속할 것을 고려하여야 한다.
임신 중 노출에 대한 전향적 코호트 등록 연구에서, 임신 1기 동안 아달리무맙을 최소 1 회 투여한 류마티스 관절염 또는 크론병이 있는 257명의 여성과 아달리무맙을 투여받지않은 류마티스 관절염 또는 크론병이 있는 120명의 여성이 등록되었다.
일차 평가변수는 주요출생결함의 발생률이었다. 주요 출생결함이 있는 한 명이상의 생존 신생아 출산으로 종료된 임신율은 아달리무맙을 투여받은 여성에서 6/69(8.7%), 치료를 받지 않은 류마티스관절염 여성에서 5/74 (6.8%) (보정하지 않은 odd ratio 1.31, 95% 신뢰구간 0.38 ‑4.52)이었고, 아달리무맙을 투여받은 크론병 16/152 (10.5%), 치료를 받지 않은 크론병 여성에서 3/32 (9.4%)(보정하지 않은 odd ratio 1.14, 95% 신뢰구간 0.31 ‑4.16)이었다. 류마티스관절염과 크론병의 베이스라인 차이를 보정한 odd ratio는 1.10 (95% 신뢰구간 0.45 ‑2.73)이었다.
이차 평가변수인 경미한 출생결함, 조기분만, 저체중, 심각한 또는 기회 감염에서 유의한 차이가 관찰되지 않았다 (보정한 odd ratio 0.84, 95% 신뢰구간 0.34, 2.05). 사산이나 악성 종양은 보고되지 않았다.
이 등록연구는 적은 표본 크기와 무작위연구 설계가 아니라는 방법론적인 한계로 데이터 해석에 영향을 줄 수 있다.
아달리무맙은 임신 중 아달리무맙으로 치료받은 여성에서 태반을 통과하여 태아의 혈청으로 들어간다. 결과적으로 이 유아들은 감염의 위험이 증가될 수 있다. 자궁에서 아달리무맙에 노출된 유아의 경우, 임신 중 이 약의 마지막 투여 후 5개월 동안 생백신 투여는 권장되지 않는다.
2) 출판된 문헌의 제한된 정보에서 모유로 분비되는 아달리무맙의 농도는 매우 낮으며, 인간의 모유 내 아달리무맙 농도는 모체 혈청 수치의 0.1%에서1% 정도이다. 경구로 섭취한 면역글로불린 G 단백질은 장내에서 단백질 분해되고 생체이용률이 낮으므로, 모유수유하는소아에 대한 아달리무맙의 전신 영향 가능성은 거의 없을 것이다. 모유수유의 발달 및 건강상 이익은 아달리무맙에 대한 수유부의 임상적 필요성과 아달리무맙 또는 수유부의 기존 상태로 인한 소아에 대한 잠재적인 유해성과 함께 고려해야 한다.
7. 소아에 대한 투여
이 약은 소아 환자에 대해 연구되지 않았으므로 추가적인 자료를 얻을 때까지 18세 미만 소아에 대한 투여는 권장되지 않는다[소아 특발성 관절염(다관절형 소아 특발성 관절염, 골부착부위염 관련 관절염), 크론병(6 ‑ 17세) 및 판상 건선 제외].
8. 고령자에 대한 투여
이 약의 용량 조절이 요구되지 않는다. 이 약의 임상시험에 참가한 환자 중 65세 이상이 9.4%이고 75세 이상이 약 2.0%였으며 이들 환자군과 더 젊은 환자군 사이에 전반적인 유효성의 차이는 관찰되지 않았다. 이 환자군에서 용량조절은 필요하지 않다.
9. 과량투여시의 처치
임상시험 중 용량제한독성은 관찰되지 않았다. 평가된 최고 용량은 10 mg/kg의 수회 정맥투여였다. 과량투여의 경우 이상사례의 증상이나 징후 또는 효과를 모니터링하고 적절한 대증적 처치를 한다.
10. 적용상의 주의사항
1) 주사방법에 대한 교육을 받은 후 환자가 자가주사할 경우, 자가주사 부위는 대퇴부 또는 복부를 포함하며 주사부위는 교대로 바꿔야 한다. 새로운 주사부위는 앞서 주사한 부위에서 최소 3 cm 정도 떨어져서 주사한다.
2) 새로운 주사부위가 약하거나, 멍들거나, 충혈되거나 딱딱한 경우에는 투여해서는 안 된다. 투여 전 변색 및 이물질에 대해 육안으로 관찰한다. 주사액은 무색투명해야 하며, 주사액이 변색 또는 불투명하거나 이물이 있을 경우 투여하지 않는다. 펜형 제품의 경우 몸체에 있는 창(window)을 통해 확인한다. 주사 전 제품을 흔들거나 떨어뜨리지 않는다.
회색 뚜껑 및 자주색 뚜껑을 주사 직전에 제거한다.
3) 프리필드시린지
주사부위를 동봉된 알콜솜으로 닦는다. 주사바늘 덮개를 벗기고, 주사부위를 부드럽게 잡아 고정한 후 피부와 45도 각도로 주사한다.
4) 펜(pen) 형
⓵ 주사부위를 동봉된 알콤솜으로 닦는다. 회색 뚜껑 및 자주색 뚜껑을 주사 직전에 제거한다. 펜의 회색부분을 한손으로 잡는다. 펜의 회색 뚜껑(1) 및 자주색 뚜껑(2)에 손이 닿지 않도록 펜의 가운데를 손으로 잡는다. 회색 뚜껑(1)이 위로 향하도록 잡는다.
다른 손으로 회색 뚜껑(1)을 즉시 당겨서 제거한다. 작은 회색 주사바늘 커버와 덮개가 함께 제거되었는지 확인하며, 뚜껑을 다시 씌우면 안 된다. 주사침에서 몇 방울의 액이 나오는 것은 정상이다.
⓶ 펜의 자주색 뚜껑(2)을 잡아당겨 벗긴 후 버린다. 자주색 작동 버튼이 노출되어 펜을 사용할 준비가 된다. 약물이 방출될 수 있으므로, 적절한 주사위치를 잡기 전까지 자주색 작동 버튼을 누르거나 뚜껑을 다시 씌우면 안 된다.
⓷ 한 손으로 주사부위의 깨끗한 피부를 아래 그림처럼 부드럽게 잡아 고정시킨다.
⓸ 펜의 흰색 끝부분을 창이 보이도록 하여 피부와 직각(90도)이 되도록 놓는다. 창으로 보여 지는 몇 방울의 기포는 정상이다.
⓹ 펜의 몸체를 잡고 주사부위에 가볍게 눌러 주사준비를 한 다음 집게손가락 또는 엄지손가락으로 주사기 윗부분의 자주색 버튼을 누른다. 바늘이 나올 때 나는 ‘딸깍’ 소리가 들리고, 바늘에 찔리는 따끔함이 느껴진다.
➅ 약 10초간 유지하여 완전히 주사되도록 한다.
⑦ 주사 동안 노란색 표시가 펜의 창내로 이동하며, 노란색 표시의 이동이 멈추면 주사가 끝난 것이다. 노란색 표시는 펜의 주사막대의 일부분이다. 노란색 표시가 창에 나타나지 않는다면, 주사막대가 적절히 이동하지 않은 것이고, 주사가 완전히 이루어지지 않은 것이다.
5) 이 약의 주사기에 다른 약물을 혼합해서는 안 된다. 사용하지 않고 남은 부분은 버린다. 일회용이며 재사용하지 않는다. 어린이의 손이 닿지 않는 곳에 보관한다.
11. 저장상의 주의사항
1) 이 약은 동결을 피하여 냉장 보관(2 ‑ 8℃)한다. 사용 전까지 포장을 뜯지 않고 용기 상자 내부에 보관한다.
2) 이 약은 냉장조건을 벗어난 경우 1회에 한하여 25℃이하에서 최대 14일간 보관할 수 있으며, 빛을 피하여 보관하여야 한다. 냉장조건을 벗어난 제품은 다시 냉장보관해서는 안되며, 14일 이내 사용하거나 폐기하여야 한다.
12. 기타
이 약은 류마티스 관절염 환자에 대한 한 건의 이중 눈가림 후 공개연장 임상시험에서 최대 60개월까지 투여되었다. 60개월까지의 투여기간 동안 류마티스 관절염의 증상과 징후를 감소시키는 효과가 유지되었고 관절손상 진행속도 감소와 신체활동기능 향상 효과가 유지되었으며, 전반적으로 안전하고 내약성이 좋았다.
13. 전문가를 위한 정보
1) 임상약리학적 정보
⓵ 일반 정보
아달리무맙은 TNF에 특이적으로 결합하여 TNF와 p55 및 p75 세포 표면 TNF 수용체의 상호작용을 차단함으로써 TNF의 생물학적 기능을 중화시킨다. TNF는 자연적으로 발생하는 사이토카인으로서 정상적인 염증 및 면역 반응에 관여한다. 또한 아달리무맙은 백혈구 이동(ELAM ‑1, VCAM ‑1 및 ICAM ‑1, IC50 1 ‑2 × 10 ‑<SUP>10</SUP>M)의 원인이 되는 부착 분자의 수치 변화와 같이 TNF에 의해 유도되거나 조절되는 생물학적 반응을 조절한다.
⓶ 약력학적 정보
이 약을 투여한 후 류마티스 관절염 환자에서 베이스라인과 비교하여 염증의 급성 단계 반응물질(C ‑반응성 단백질[C ‑reactive protein; CRP]과 적혈구 침강속도[erythrocyte sedimentation rate; ESR] 과 혈중 사이토카인[IL ‑6]) 수치가 급격히 감소하였다. 또한 소아 특발성 관절염, 크론병, 궤양성 대장염 및 화농성 한선염 환자에서 CRP 수치 감소뿐만 아니라 크론병 환자의 대장에서 인간 백혈구 항원(human leukocyte antigen; HLA ‑DR) 및 골수세포형과산화효소(myeloperoxidase; MPO)와 같은 염증 표지자와 TNF가 유의하게 감소하였다. 연골 손상의 원인이 되는 조직 리모델링을 일으키는matrix metalloproteinase(MMP ‑1 및 MMP ‑3)의 혈중 수치 또한 이 약의 투여 후 감소하였다. 류마티스 관절염, 건선성 관절염, 강직성 척추염 환자는 경증부터 중등도의 빈혈 및 림프구 수 감소뿐 아니라 호중구와 혈소판 수 증가를 종종 경험한다. 이 약을 투여 받은 환자는 대개 만성 염증의 혈액학적 징후에서 개선을 경험하였다.
그림 1: 농도 ‑효과 연관성
⓷ 약동학적 정보
흡수
건강한 성인 대상자 59명에게 아달리무맙 40 mg을 단회 피하 투여한 아달리무맙의 흡수 및 분포는 느리며 투여 후 약 5일 뒤에 평균 최대 혈중 농도에 도달하였다. 3건의 시험에서 40 mg 단회 피하 투여 후 추정한 아달리무맙의 절대 생체이용률은 평균 64%였다.
분포 및 배설
아달리무맙의 단일 투여 약동학은 0.25 ‑10 mg/kg 범위의 정맥 내 투여에 대한 여러 시험에서 확인되었다. 4.7 ‑6.0 L 범위의 분포 용적(distribution volume; Vss)은 아달리무맙이 혈관과 혈관 외액 사이에 거의 비슷하게 분포함을 나타내었다. 아달리무맙은 일반적으로 12 mL/h 이하의 청소율로 천천히 배설된다. 평균 최종 단계 반감기는 약 2주였으며 범위는 10 ‑20일이었다. 청소율 및 반감기는 시험된 투여량 범위에서 상대적으로 변화가 없었고 최종 반감기는 IV 투여 및 SC 투여에서 비슷하였다. 여러 류마티스 관절염환자 활액에서의 혈중 아달리무맙 농도 범위는 31 ‑96%였다.
정상상태(steady ‑state)에서의 약동학
류마티스 관절염 환자에게 격주로 아달리무맙 40 mg을 SC 투여한 후의 반감기를 근거로 아달리무맙의 축적을 예측할 수 있었으며 평균 정상상태 최저 농도는 각각 약 5 mcg/mL(메토트렉세이트[MTX] 병용 투여하지 않은 경우) 및 8 ‑9 mcg/mL (MTX 병용 투여한 경우)였다. 정상상태 혈중 아달리무맙 최저 수치는 격주 및 매주 SC 투여한 20, 40 및 80 mg의 투여량에 따라 거의 비례적으로 증가하였다. 2년 이상의 장기간 투여 시험에서 시간에 따른 청소율의 변화는 확인되지 않았다.
건선 환자의 경우 메토트렉세이트의 병용 투여 없이 아달리무맙 40 mg 격주 투여 동안 평균 정상상태 최저 농도는 5 mcg/mL였다.
0주차에 아달리무맙 160mg을 투여 받은 후 2주차에 80 mg을 투여 받은 화농성 한선염 환자의 혈중 아달리무맙 최저 농도는 2주차와 4주차에 약 7 ‑8 mcg/mL에 도달하였다. 12주차부터 36주차까지의 평균 정상상태 최저 농도는 매주 아달리무맙 40 mg 투여 동안 약 8 ‑10 mcg/mL였다.
포도막염 환자의 경우 0주차에 도입용량인 아달리무맙 80 mg을 투여하고 1주차부터 격주마다 아달리무맙 40 mg을 투여했을 때 평균 정상상태 농도는 약 8 ‑10 mcg/mL였다.
집단 약동학과 약동학/약력학 모델링 및 시뮬레이션 결과, 매주 40 mg의 아달리무맙 투여와 비교하여 격주 80 mg 투여한 환자의 아달리무맙 노출 및 효능이 유사한 것으로 예측하였다(성인 류마티스 관절염, 화농성 한선염, 궤양성 대장염, 크론병 또는 건선환자 및 40kg 이상의 소아 크론병 환자).
1,200명 이상 환자 데이터의 집단 약동학 분석을 통해 메토트렉세이트의 병용 투여가 아달리무맙의 겉보기 청소율(apparent clearance; CL/F)에 본질적인 영향을 미친다는 것이 확인되었다(‘5. 상호작용’ 참조). 예상한 바와 같이 체중 증가 및 항 ‑아달리무맙 항체의 존재 하에 아달리무맙의 겉보기 청소율이 높아지는 경향이 있었다.
보다 중요하지 않은 다른 요인들도 확인하였다. 권장된 투여량보다 낮은 투여량을 받은 환자, 높은 류마티스인자를 가진 환자 또는 CRP 농도가 높은 환자의 겉보기 청소율은 높을 것으로 예측되었다. 이러한 요인은 임상적으로 중요할 가능성이 낮다.
비방사선학적 축성 척추관절염 환자에게 격주마다 아달리무맙 40 mg을 피하투여한 후 68주차의 평균(±SD) 최저 정상상태 농도는 8.0 ± 4.6 μg/mL였다.
크론병 환자의 경우, 0주차에 유도용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 2주차와 4주차의 평균 혈중 아달리무맙 최저 수치는 약 12 mcg/mL였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 크론병 환자에서 24주차와 56주차의 평균 정상상태 최저 수치는 약 7 mcg/mL로 관찰되었다.
궤양성 대장염 환자의 경우, 0주차에 유도용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 유도 기간 동안의 혈중 아달리무맙 최저 농도는 약 12 mcg/mL에 도달하였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 궤양성 대장염 환자에서의 평균 정상상태 최저 수치는 약 8 mcg/mL로 관찰되었다.
베체트병이 있는 일본 환자의 경우, 0주차에 도입용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 유도 기간 동안의 혈중 아달리무맙 최저 농도는 약 13 mcg/mL에 도달하였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 장내 베체트병이 있는 일본 환자에서의 평균 정상상태 최저 수치는 약 9 mcg/mL로 관찰되었다.
특수 집단
특수한 집단에서의 약동학은 집단 약동학 분석을 사용하여 조사하였다.
고령 환자
연령은 아달리무맙의 겉보기 청소율에 대해 최소한의 영향을 미치는 것으로 나타났다. 집단 분석에서 40 ‑ 65세 환자(n=850) 및 ≧ 65세 환자(n=287)의 평균 체중을 보정한 청소율은 각각 0.33 및 0.30mL/h/kg이었다.
소아 환자
다관절형 소아 특발성 관절염이 있는 4 ‑ 17세의 환자에게 격주마다 아달리무맙 24 mg/m2(최대 40 mg)를 피하투여한 후 평균 최저 정상상태(20 ‑48주차에 측정한 수치) 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여했을 때 5.6 ± 5.6 μg/mL (102% CV), 메토트렉세이트를 병용 투여하지 않았을 때 10.9 ± 5.2 μg/mL (47.7% CV)였다. 20 mg 아달리무맙을 격주마다 피하투여한 30 kg인 환자에서 메토트렉세이트를 병용 투여하거나 병용 투여하지 않았을 때 평균 정상상태 혈중 아달리무맙 농도는 각각 6.8 μg/mL 및 10.9 μg/mL였다. 40 mg 아달리무맙을 격주마다 피하투여한 ≧30 kg인 환자에서 메토트렉세이트를 병용 투여하거나 병용 투여하지 않았을 때 평균 정상상태 혈중 아달리무맙 농도는 각각 6.6 μg/mL 및 8.1 μg/mL였다. 다관절형 소아 특발성 관절염이 있는 2세이상 4세미만 또는 ≧4세의 15 kg인 환자에게 아달리무맙 24 mg/m2를 투여했을 때의 평균 최저 정상상태 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여하지 않은 경우 6.0 ± 6.1 μg/mL (101% CV), 메토트렉세이트를 병용 투여한 경우 7.9 ± 5.6 μg/mL (71.2% CV)였다.
골부착부염 관련 관절염 환자에게 24 mg/m2(최대 40 mg)를 격주마다 피하투여한 후 평균 최저 정상상태(24주차에 측정한 수치) 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여하지 않은 경우 8.8 ± 6.6 μg/mL, 메토트렉세이트를 병용 투여한 경우 11.8 ± 4.3 μg/mL였다.
중등증부터 중증의 활동성 크론병 소아 환자에서의 공개 연장 시험 아달리무맙 유도용량은 0주차와 2주차에 체중 절단점(cut ‑off)인 40 kg에 따라 각각 160/80 mg 또는 80/40 mg이었다. 4주차에 환자는 체중에 따라 표준용량(격주 40/20 mg) 또는 저용량(격주 20/10 mg) 유지 투여군에 1:1의 비율로 무작위 배정되었다. 4주차에 도달한 평균(±SD) 혈중 아달리무맙 최저 농도는 ≧40 kg인 환자(160/80mg)의 경우 15.7 ± 6.6 μg/mL, 40 kg인 환자(80/40 mg)의 경우 10.6 ± 6.1 μg/mL였다.
무작위 배정된 치료를 지속한 환자에서 52주차의 평균(±SD) 아달리무맙 최저 농도는 표준용량 투여군의 경우 9.5 ± 5.6 μg/mL, 저용량 투여군의 경우 3.5 ± 2.2 μg/mL였다. 52주 동안 아달리무맙 격주 투여를 지속한 환자에서 평균 최저 농도는 유지되었다. 격주에서 매주 투여 요법으로 투여량을 증량한 환자의 경우 52주차의 평균(±SD) 아달리무맙 혈중 농도는 각각 15.3 ± 11.4 μg/mL(매주 40/20 mg) 및 6.7 ± 3.5 μg/mL(매주 20/10 mg)였다.
만성 판상형 건선이 있는 소아 환자에게 격주로 아달리무맙 0.8 mg/kg(최대 40 mg)을 피하투여한 후 평균 ±SD 정상상태 아달리무맙 최저 농도는 약 7.4 ± 5.8 μg/mL (79% CV)였다.
성별
환자의 체중을 보정한 후 성별과 관련된 약동학 차이는 관찰되지 않았다.
인종
인종간 면역글로불린 청소율의 차이는 예상되지 않는다. 코카시안이 아닌 인종의 제한된 데이터로부터, 아달리무맙에 대한 중요한 약동학적 차이는 관찰되지 않았다.
간장애 및 신장애 환자
간 또는 신장 기능 장애가 있는 환자에서 유효한 약동학 데이터가 없다.
질병 상태
건강한 지원자와 류마티스 관절염 환자는 유사한 아달리무맙 약동학을 보였다.
2) 임상시험 정보
⓵ 성인
류마티스 관절염
모든 류마티스 관절염 임상시험에서 3,000명 이상의 환자를 대상으로 이 약을 평가하였다. 이 약의 안전성 및 유효성은 5건의 무작위배정, 이중눈가림 된 시험에서 평가하였다. 일부 환자는 60개월 이상의 기간 동안 이 약을 투여 받았고, 일부는 최대 120개월의 기간 동안 이 약을 투여 받았다. 이 약 40 mg/0.8 mL과 비교한 이 약 40 mg/0.4 mL에 대한 투여 부위 통증은 2건의 무작위 배정, 활성대조, 단일 눈가림, 두 기간 교차시험으로 평가하였다.
RA 시험 I (DE009)은 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 271명을 대상으로 평가하였으며, 이 환자들은 1 ‑4개의 DMARDs(disease ‑modifying anti ‑rheumatic drug),(예: 히드록시클로로퀸, 경구 또는 주입 가능한 금, 아자티오프린, D ‑페니실라민, 설파살라진) 치료에 실패 이력이 있고 매주 12.5 ‑25 mg (메토트렉세이트 내약성이 없는 경우 10 mg)의 메토트렉세이트에 대해 효능이 불충분하며 메토트렉세이트 투여량을 매주 10 ‑25 mg으로 일정하게 유지하였다. 환자는 6개 이상의 종창 관절 및 9개 이상의 압통 관절이 있었으며 ACR 기준에 따라 류마티스관절염로 진단되었다. 이들은 24주 동안 격주마다 20, 40 또는 80 mg의 이 약이나 위약을 투여 받았다.
RA 시험 II (DE011)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 544명을 대상으로 평가하였으며, 이들은 최소 하나의 DMARD(예: 메토트렉세이트, 설파살라진, 히드록시클로로퀸, 경구 또는 주입 가능한 금, D ‑페니실라민, 아자티오프린) 치료에 실패한 이력이 있었다. 환자는 10개 이상의 종창 관절 및 12개 이상의 압통 관절이 있었으며 ACR 기준에 따라서도 진단되었다. 이들은 26주 동안 이 약 20 mg 또는 40 mg을 위약과 격주로 번갈아 피하 투여 받거나 매주 피하 투여 받았다. 위약은 동일한 기간 동안 매주 투여 받았다.
RA 시험 III (DE019)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 619명을 대상으로 평가하였으며, 매주 12.5 ‑25 mg (메토트렉세이트 내약성이 없는 경우 10 mg)의 메토트렉세이트에 대해 효능이 불충분하며 메토트렉세이트 투여량을 매주 12.5 ‑25 mg으로 일정하게 유지하였다. RA 시험 I과는 다르게, RA 시험 III의 환자는 메토트렉세이트 외의 DMARD 치료 실패 이력이 필요하지 않았다. 환자는 6개 이상의 종창 관절 및 9개 이상의 압통 관절이 있었으며 ACR 기준에 따라 류마티스 관절염으로 진단되었다. 이 시험에는 3개의 군이 있었다. 첫 번째 군은 52주 동안 매주 위약을 투여 받았다. 두 번째 군은 52주 동안 매주 이 약 20 mg 을 투여 받았다. 세 번째 군은 이 약 40 mg과 위약을 격주로 번갈아 투여 받았다. 첫 52주 동안 투여를 완료한 후 457명의 환자는 최대 60개월 동안 격주로 이 약 40 mg/메토트렉세이트를 투여 받는 공개 연장 시험에 등록되었다. 첫 52주 동안 투여를 완료한 후 457명의 환자는 최대 10년 동안 격주로 이 약 40mg /메토트렉세이트를 투여 받는 공개 연장 시험에 등록되었다.
RA 시험 IV (DE031)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 636명을 평가하였다. 이 환자들은 최소 3개월 동안 류마티스 관절염 진단에 대한 ACR 기준을 충족했으며 최소 6개의 종창 관절 및 9개의 압통 관절을 가지고 있었다. 최소한 28일 동안 치료가 안정적이었을 경우, DMARD ‑I 또는 기존의 류마티스 치료를 유지하는 것이 허용되었다. 환자는 24주 동안 이 약 격주 40 mg투여군 또는 위약군에 무작위 배정되었다.
RA 시험 V (DE013)는 중등증에서 중증의 활동성 초기 류마티스 관절염(평균 질병 기간이 9개월 미만)이 있는 메토트렉세이트 투여 이력이 없는 성인 환자 799명을 대상으로 평가하였다. 이 시험에서는 104주 동안 류마티스 관절염에서 류마티스 관절염의 징후 및 증상을 감소시키고 관절 손상의 진행 속도를 줄이는 데 대한 이 약 40 mg 격주/메토트렉세이트 병용 치료, 이 약 40 mg 격주 단일 치료 및 메토트렉세이트 단일 치료의 효능을 평가하였다. 첫 104주 동안 투여를 완료한 후 497명의 환자는 최대 10년 동안 이 약 격주로 40 mg을 투여 받는 공개 연장 시험에 등록되었다.
RA 시험 VI 및 VII는 각각 중등증에서 중증의 활동성 류마티스 관절염이 있는 ≧ 18세 환자 60명을 대상으로 평가하였다. 등록된 환자는 당시 이 약 40 mg/0.8 mL를 투여 받는 중이며 평균 투여 부위 통증이 최소 3cm (0 ‑10cm VAS)인 환자, 또는 이 약 40 mg/0.8 mL를 투여 받기 시작한 생물학적 치료 이력이 없는 환자였다. 환자는 이 약 40 mg/0.8 mL 또는 이 약 40 mg/0.4 mL 단일 투여군으로 무작위 배정된 후 다음 파트에 교차 투여된다.
RA 시험 I ‑V의 결과는 ACR 반응 기준을 사용하여 류마티스 관절염에서 개선을 보인 환자 비율로 표현하였다. RA 시험 I, II 및 III의 일차 평가변수와 RA 시험 IV의 이차 평가변수는 24주 또는 26주차에 ACR 20 반응에 도달한 환자의 비율이었다. RA 시험 V의 일차 평가변수는 52주차에 ACR 50 반응에 도달한 환자 비율이었다. RA 시험 III 및 V의 52주차에서 추가적인 일차 평가변수는 질병 진행의 지연이었다(엑스레이 결과로 확인). RA 시험 III 또한 삶의 질 변화를 일차 평가변수로 설정하였다. RA 시험 VI 및 VII의 일차 평가변수는 0 ‑10 cm VAS로 측정한 투여 직후의 투여 부위 통증이었다.
임상 반응
RA 시험 I, II 및 III
이 약을 투여 받은 환자 중 ACR 20, 50 및 70 반응에 도달한 환자 비율은 3건의 시험 모두에서 일관적이었다. 3건의 시험 결과는 표 3에 요약한다.
표 3: 위약대조 시험에서의 ACR 반응(환자 비율)
a RA 시험 I 24주차, RA 시험 II 26주차, RA 시험 III 24주차 및 52주차
b 40mg 이 약 격주 투여
* p0.01, ACR 20, 50, 70에 대한 모든 시점에서 이 약 vs. 위약
RA 시험 II에서 매주 이 약 40 mg을 투여 받은 환자 또한 6개월차에 통계적으로 유의하게 ACR 20, 50 및 70 반응률에 각각 53.4%, 35.0% 및 18.4% 도달하였다.
RA 시험 III에 대한 ACR 반응 기준의 구성요소 결과는 표 4에 제시되었다. 아래에 제시한 결과는 일반적으로 수행된 각 시험을 대표하는 것이다.
ACR 반응률 및 모든 ACR 반응 기준의 개선은 104주차까지 유지되었다. RA 시험 III에서 2년 동안 이 약 투여군의 24%가 6개월 동안 ACR 70 반응을 유지한 것으로 정의되는 주요 임상 반응에 도달하였다. ACR 반응은 RA 시험 III의 공개 연장 시험에서 이 약 치료를 지속한 환자 중 비슷한 비율의 환자에서 최대 60개월 동안 유지되었다.
RA 시험 III의 공개 연장에서 ACR 반응자 중 대다수의 환자가 최대 10년간의 추적조사 시 반응을 유지했다. 격주 이 약 40 mg 투여군으로 무작위 배정된 환자 207명 중 114명은 5년 동안 격주로 이 약 40 mg을 지속적으로 투여 받았다. 이들 중 86명(75.4%)은 ACR 20 반응, 72명(63.2%)은 ACR 50 반응, 41명(36%)은 ACR 70 반응에 도달하였다. 207명의 환자 중 81명은 10년 동안 이 약 40 mg을 지속적으로 투여 받았다. 이들 중 64명(79.0%)은 ACR 20 반응, 56명(69.1%)은 ACR 50 반응, 43명(53.1%)은 ACR 70 반응에 도달하였다.
표 4: RA 시험 III에서의 ACR 반응 구성요소
RA 시험 III에서 24주차에 ACR 20 반응에 도달한 환자의 84.7%는 52주차에도 반응을 유지하였다. 다음의 그림은 시험 III 및 II에서이 약에 대한 ACR 20 반응의 지속성을 나타낸다.
RA 시험 IV
이 약과 함께 표준치료를 투여 받은 환자의 ACR 20 반응은 위약과 함께 표준치료를 투여 받은 환자에 비해 통계적으로 유의하게 우수하였다(p0.001). 이 약과 다른 DMARD의 병용 투여와 관련된 고유의 이상사례는 관찰되지 않았다.
RA 시험 I ‑IV에서 이 약을 투여 받은 환자는 위약을 투여 받은 환자보다 더 빠르고 자주 ACR 20, 50 및 70 반응에 도달하였다. RA 시험 I에서 1주차(첫 시험 방문)의 ACR 20 반응에 대해 이 약을 투여 받은 환자(26.0%)와 위약을 투여 받은 환자(5.0%) 사이에 통계적으로 유의한 차이가 있었다. ACR 20 반응에 대한 통계적으로 유의한 차이는 RA 시험 II, III 및 IV의 2주차(첫 시험 방문)에 이 약을 투여 받은 환자(각각 36.4%, 29.1% 및 33.7%)와 위약을 투여 받은 환자(각각 7.3%, 13.0% 및 8.6%) 사이에서도 확인되었다. 첫 ACR 50 및 70 반응까지의 시간은 4건의 시험 모두에서 유사한 패턴으로 나타났다.
메토트렉세이트를 병용 투여하지 않은 일부 환자에서 매주 40 mg으로 이 약의 투여 빈도를 증가시킴에 따라 추가 이익이 유도될 수 있다. 이는 불완전한 반응을 보인 환자의 투여 빈도를 격주 40 mg에서 매주 40mg으로 증가시킨 장기간 공개 연장 시험에서 확인되었다.
RA 시험 V
메토트렉세이트의 투여 이력이 없는 초기 류마티스 관절염 환자를 대상으로 한 RA 시험 V에서, 이 약과 메토트렉세이트의 병용 치료는 52주차에 메토트렉세이트 단일 치료 및 이 약 단일 치료와 비교하여 더 빠르고 유의하게 큰 ACR 반응을 유발했으며 이 반응은 104주까지 지속되었다(표 5참조).
52주차에 ACR 반응 기준의 모든 개별적인 구성요소는 이 약/메토트렉세이트 치료로 인해 개선되었으며 개선은 104주까지 유지되었다.
2년간의 시험 동안, 이 약/메토트렉세이트 병용 치료를 받은 환자 중 48.5%가 주요 임상 반응(6개월 연속 ACR 70)에 도달했으며, 이에 비해 메토트렉세이트 단일 치료를 받은 환자는 27.2% (p0.001), 이 약 단일 치료를 받은 환자는 24.5% (p0.001)가 주요 임상 반응에 도달하였다.
표 5: RA 시험 V에서의 ACR 반응(환자 비율)
a 주요 임상 반응은 6개월 연속 ACR 70 반응에 도달한 것으로 정의한다.
b p0.05, ACR 20에 대한 이 약/MTX vs. MTX
p0.001, ACR 50, 70 및 주요 임상 반응에 대한 이 약/MTX vs. MTX
c p0.001, 이 약/MTX vs. 이 약
RA 시험 V의 공개 연장시험에서 ACR 반응률은 최대 10년간의 추적조사 시에 유지되었다. 이 약 격주 40 mg 투여군으로 무작위 배정된 환자 542명 중 170명은 10년 동안 이 약 격주 40 mg 투여를 지속하였다. 이들 중 154명(90.6%)은 ACR 20 반응, 127명(74.7%)은 ACR 50 반응, 102명(60.0%)은 ACR 70 반응에 도달하였다. 52주차에 이 약/메토트렉세이트 병용 치료를 받은 환자 중 42.9%가 임상적 관해(질병 활성도 점수[Disease Activity Score; DAS28] ‑CRP 2.6)에 도달하였고, 이에 비해 메토트렉세이트 단일 치료를 받은 환자는 20.6%, 이 약 단일 치료를 받은 환자는 23.4%가 임상적 관해에 도달하였다. 이 약/메토트렉세이트 병용 치료는 최근에 중등증부터 중증의 류마티스 관절염을 진단 받은 환자가 낮은 질병 상태에 도달하는 데 있어 메토트렉세이트(p0.001) 및 이 약 단일 치료(p0.001)보다 통계적 및 임상적으로 우수하였다(표 6참조). 기존에 이 약 단일 치료 또는 이 약/메토트렉세이트 병용 치료로 무작위 배정되어 공개 연장 시험에 등록된 342명의 대상자 중 171명이 10년 동안의 이 약 치료를 완료하였다. 이들 중 109명(63.7%)이 10년차에 질병 관해를 보고하였다.
표 6: RA 시험 V에서의 DAS28 반응
a p0.001, 이 약/메토트렉세이트 vs. 메토트렉세이트
b p0.001, 이 약/메토트렉세이트 vs. 이 약
방사선학적 반응
RA 시험 III에서 이 약을 투여 받은 환자의 류마티스 관절염 평균 지속 기간은 약 11년이었으며 구조적 관절 손상은 방사선 촬영을 통해 평가하였고 수정된 전체 샤프 점수(modified total Sharp score; TSS) 및 구성요소, 미란(erosion) 점수 및 관절 간격 협착(joint space narrowing score; JSN)의 변화로 결과를 표시하였다. 손/손목 및 발의 앞부분의 방사선 사진은 베이스라인, 6개월 및 12개월에 판독하였다. 12개월 결과는 표 7에 제시한다. 6개월 때 TSS 및 미란 점수의 변화에서 통계적으로 유의한 차이가 관찰되었고 12개월 때 유지되었다. 52주차에 이 약/메토트렉세이트를 투여 받은 환자는 MTX만 단독 투여 받은 환자보다 방사선학적 진행이 더딘 것으로 확인되었다.
표 7: MTX를 병용 투여한 RA 시험 III에서 12개월간의 방사선학적 변화
a 격주 40mg 투여
b ranked ANCOVA 분석에 기초함
RA 시험 III의 공개 연장 시험에서 특정 용량의 이 약을 투여 받은 기존의 환자 중 77%는 2년차에 방사선 촬영으로 평가하였다. TSS로 측정한 결과, 환자는 구조적 손상의 억제를 유지했다. 54%의 환자는 구조적 손상의 진행이 없었다(0점 이하의 TSS 변화로 정의). 격주로 이 약 40 mg을 기존에 투여 받은 환자 중 55%는 5년차에 방사선 촬영으로 평가하였다. 환자의 구조적 손상 억제는 지속적이었으며 50%는 구조적 손상의 진행이 없었다(0점 이하의 TSS 변화로 정의).
RA 시험 III의 공개 연장 시험에서 구조적 손상의 진행률 감소는 환자의 하위군에서 8 ‑10년 동안 유지되었다. 격주로 이 약 40 mg을 기존에 투여 받은 환자 207명 중 81명의 환자를 8년차에 방사선 촬영으로 평가하였다. 이들 중 48명의 환자는 구조적 손상의 진행이 없었다(베이스라인으로부터 mTss 변화가 0.5 이하). 격주로 이 약 40 mg을 기존에 투여 받은 환자 207명 중 79명의 환자를 10년차에 방사선 촬영으로 평가하였다. 이들 중 40명의 환자는 구조적 손상의 진행을 보이지 않았다(베이스라인으로부터 mTss 변화가 0.5 이하).
RA 시험 V에서의 구조적 관절 손상은 RA 시험 III과 동일하게 평가하였다. 방사선학적 진행의 억제(TSS, 미란 점수 및 JSN의 변화로 평가)는 52주차와 104주차 모두, 메토트렉세이트 또는 이 약 단일 투여군보다 이 약/메토트렉세이트 병용 투여군에서 높게 관찰되었다(표 8참조).
표 8: RA 시험 V에서 52주차의 방사선학적 평균 변화
a p0.001, 52주차 및 104주차에서 이 약/MTX vs. MTX
b p0.01, 52주차 이 약/MTX vs. 이 약
p0.001, 104주차 이 약/MTX vs. 이 약
52주 및 104주간의 투여 이후, 질병 진행이 없는(베이스라인으로부터 수정된 전체 샤프 점수의 변화 ≦0.5점) 환자의 비율은 이 약/메토트렉세이트 병용 치료군(각각 63.8% 및 61.2%)이 메토트렉세이트 단일 치료군(각각 37.4%, 33.5%, p0.001)과 이 약 단일 치료군(각각 50.7%, p0.002 및 44.5%, p0.001)에 비해 유의하게 높았다.
RA 시험 V의 공개 연장 시험에서 베이스라인으로부터 10년차까지 수정된 전체 샤프 점수의 평균 변화는 메토트렉세이트 단일 치료군, 이 약 단일 치료군 및 이 약/메토트렉세이트 병용 치료군으로 기존에 무작위 배정된 환자에서 각각 10.8점, 9.2점 및 3.9점이었다. 방사선학적 진행이 없는 환자의 비율은 각각 31.3%, 23.7% 및 36.7%였다.
삶의 질 및 신체적 기능
5건의 적절하고 잘 제어된 시험 모두에서 건강 평가 설문조사(Health Assessment Questionnaire; HAQ)의 장애 척도를 사용하여 건강 관련 삶의 질을 평가하였고, 이는 RA 시험 III에서 사전에 명시된 52주차의 일차 평가변수였다. 4건의 시험 모두에서 이 약의 모든 투여/계획은 베이스라인으로부터 6개월차까지 HAQ의 장애 척도에 대해 위약보다 통계적으로 유의하게 큰 개선을 보였다. RA 시험 III에서 베이스라인으로부터 52주차까지의 HAQ 평균(CI) 개선은 이 약/메토트렉세이트 환자의 경우 ‑0.60 ( ‑0.65, ‑0.55)이었고 위약/메토트렉세이트 환자(p0.001)의 경우 ‑0.25 ( ‑0.33, ‑0.17)였다. 이 약/메토트렉세이트를 투여 받은 환자 중 63%는 시험의 이중눈가림 부분에서 52주차에 0.5 이상의 HAQ 개선에 도달하였다. 이 환자들 중 82%는 104주차까지 개선이 유지되었으며 비슷한 비율의 환자가 공개 연장시험의 260주차(5년)까지 반응이 유지되었다. 신체적 기능이 개선되고 치료를 지속한 대부분의 환자는 공개 연장 시험의 520주차(10년)까지 개선이 유지되었다.
적절하게 잘 제어된 4건의 시험 모두에서 일반적인 건강 관련 삶의 질을 평가하기 위해 약식 건강 조사(SF ‑36) 또한 사용하였다. 4건의 시험 모두에서 이 약의 모든 투여/계획은 베이스라인으로부터 6개월차까지의 SF ‑36 신체적 구성요소 요약 점수에 대해 통계적으로 유의하게 큰 개선을 보였고 이는 RA 시험 III의 52주차에서도 유지되었다. SF ‑36의 평균 개선 또한 156주차(36개월)의 측정 종료 시점까지 유지되었다. 시험 II 및 IV의 SF ‑36 정신적 구성요소 요약 점수 또한 6개월차에 이 약이 위약보다 통계적으로 유의하게 높았다. 이 약을 40mg 격주투여한 4건의 시험 모두에서 베이스라인으로부터 6개월차까지 SF ‑36의 통증 및 활력 영역 점수는 위약과 비교하여 이 약을 투여 받은 환자에서 통계적으로 유의하게 큰 개선을 보였다. 이러한 결과는 분석한 3건의 시험 모두 6개월차에 피로가 통계적으로 유의하게 감소함을 나타낸 만성질환 치료의 기능적 평가(functional assessment of chronic illness therapy; FACIT) 점수로 뒷받침되었으며 RA 시험 III의 52주차에서도 유지되었다.
RA 시험 V에서 HAQ 장애 척도 및 SF ‑36의 신체적 구성요소는 52주차에 메토트렉세이트 단일 치료군 및 이 약 단일 치료군 모두와 비교하여 이 약/메토트렉세이트 병용 치료군에서 더욱 개선되었다(p0.001). 이 개선은 104주차까지 유지되었다. 공개 연장시험을 완료한 250명의 대상자에서 신체적 기능의 개선은 10년의 치료 동안 유지되었다.
투여 부위 통증
통합 교차 RA 시험 VI 및 VII의 경우 아달리무맙 40mg/0.8mL과 아달리무맙 40mg/0.4mL 사이에서 투여 직후 투여 부위 통증에 대해 통계적으로 유의한 차이가 관찰되었다(평균 VAS 3.7cm vs. 1.2cm, 0 ‑10cm 척도, p0.001). 이는 투여 부위 통증의 84% 중앙값 감소를 나타낸다.
건선성 관절염
2건의 무작위배정, 이중눈가림, 위약 대조 시험에서 건선성 관절염 환자 413명을 대상으로 이 약 안전성과 유효성을 평가하였다. PsA 시험 I (M02 ‑518)에는 중등증부터 중증의 활동성 건선성 관절염(>3개의 종창 관절 및 >3개의 압통 관절)이 있는 성인 환자 313명이 등록되었으며 이들은 다음의 유형 중 하나에서 NSAID 치료에 대해 부적절한 반응을 보였다: (1) 원위지절간관절(distal interphalangeal; DIP) 관련(N=23), (2) 다관절형 관절염(류마티스 결절이 없고 건선이 존재)(N=210), (3) 단절성 관절염(N=1), (4) 비대칭성 건선성 관절염(N=77), 또는 (5) 강직성 척추염 유사(N=2). 등록 시 MTX 치료(>1개월 동안 안정적 용량 ≦30 mg/주) 중이었던 환자(313명 중 158명)는 동일 용량으로 메토트렉세이트를 계속 투여 받을 수 있었다. 24주의 이중눈가림 기간 동안 격주로 이 약 40 mg 또는 위약을 투여 받았다. 12주간 수행한 PsA 시험 II (M02 ‑570)는 DMARD 치료에 부적절한 반응을 보인 100명의 환자를 대상으로 하였다. 두 시험의 완료 후 383명의 환자는 공개 연장시험에 등록되어 격주로 40 mg의 이 약을 투여 받았다.
ACR 및 PASI 반응
위약과 비교하여, 이 약의 치료는 질병 활성도 측정에서 개선을 보였다(표 9 및 10 참조). 이 약을 투여 받은 건선성 관절염 환자 중, 첫 방문 시점(2주차)에서 일부 환자의 임상적 반응이 명백하였다. 비록 단절성 관절염 및 강직성 척추염 유사 하위유형에는 극소수의 환자가 등록되었지만 PsA의 각 하위유형 환자에서도 비슷한 반응이 관찰되었다. 베이스라인에서 메토트렉세이트치료를 병용 투여 받거나 병용 투여 받지 않은 환자에서도 반응이 비슷하였다.
최소 3% 체표면적(body surface area; BSA)이 건선과 관련된 환자에서 건선 면적 및 중증도 지수(Psoriatic Area and Severity Index; PASI) 반응을 평가하였다. 24주차에 PASI가 75% 또는 90% 개선에 도달한 환자의 비율은 이 약 투여군(N=69)에서 각각 59%와 42%였고, 이에 비해 위약군(N=69)에서는 각각 1%와 0%였다(p0.001). PASI 반응은 첫 방문 시점(2주차)에 일부 환자에서 명백하였다. 베이스라인에서 메토트렉세이트 치료를 병용 투여 받거나 병용 투여 받지 않은 환자에서도 반응이 비슷하였다.
표 9: 위약대조 건선성 관절염 시험에서의 ACR 반응(환자 비율)
a 이 약과 위약 사이의 모든 비교에 대해 p0.001
b 이 약과 위약 사이의 모든 비교에 대해 p0.05
N/A 해당 없음
표 10: 건선성 관절염 PsA 시험 I에서의 질병 활성도 구성요소
* 중앙값 변화를 기초로 이 약 vs. 위약 비교에 대해 p0.001
a 0 ‑78 척도
b 0 ‑76 척도
c 시각적 아날로그 척도. 0점 = 최고, 100점 = 최악
d 건강 평가 설문조사의 장애 척도. 0점 = 최고, 3점 = 최악. 옷 입기/치장하기, 일어나기, 먹기, 걷기, 뻗기, 잡기, 위생 유지 및 일상 활동 유지 등을 수행할 수 있는 환자의 능력 측정
e 정상 범위: 0 ‑2.87mg/L
최대 136주 동안의 공개 연장시험에서 ACR 반응은 유지되었다.
건선성 관절염 시험에서 방사선학적 변화를 평가하였다. 손, 손목 및 발의 방사선 사진은 환자가 이 약 또는 위약을 투여 받은 이중눈가림 기간의 베이스라인과 24주차, 그리고 모든 환자가 공개 연장 시험의 이 약을 투여 받은 48주차에 수집하였다. 원위지절간관절을 포함한 수정된 전체 샤프 점수(modified Total Sharp Score; mTss)(즉, 류마티스 관절염에서 사용한 TSS와 동일하지 않음)를 사용하였다.
이 약 치료는 위약 치료와 비교하여 말초 관절 손상의 진행률을 감소시켰고, 이는 베이스라인으로부터 mTss 점수(평균 ± SD) 변화로 측정하였으며 위약군의 경우 0.8 ± 2.5 (24주차), 이 약 투여군의 경우 0.0 ± 1.9 (48주차)였다(p0.001).
베이스라인으로부터 48주차까지 방사선학적 진행이 없는 이 약 투여군(n=102) 중, 84%는 144주간의 치료 동안 방사선학적 진행을 보이지 않았다.
삶의 질 및 신체적 기능
이 약을 투여 받은 환자는 24주차에 위약과 비교하여 HAQ 및 약식 건강 조사(SF ‑36) 평가 결과 신체적 기능에 있어서 통계적으로 유의한 개선이 확인되었다. 개선된 신체적 기능은 최대 136주차의 공개 연장 시험동안 지속되었다.
축성 척추관절염 임상시험
강직성 척추염
활동성 강직성 척추염(ankylosing spondylitis; AS)이 있는 성인 환자 393명을 대상으로 2건의 무작위배정, 24주 이중눈가림, 위약대조 시험에서 이 약 40 mg의 격주 투여에 대한 안전성과 유효성을 평가하였다. 2건 중 규모가 큰 시험(AS 시험 I 또는 M03 ‑607)에는 활동성 강직성 척추염이 있고 기존의 치료에 부적절한 반응을 보인 성인 환자 315명이 등록되었다. 활동성 강직성 척추염은 다음의 3가지 기준 중 최소 2가지를 충족하는 것으로 정의한다: (1) 강직성 척추염 질병 활성도 지수(Bath AS disease activity index; BASDAI] 점수 ≧4 cm, (2) 전체 요통에 대한 시각적 아날로그 점수(visual analog score; VAS) ≧40 mm, (3) 조조 강직 ≧1시간. 눈가림 기간 후 공개 연장시험 기간 동안 환자는 최대 236주의 추가 기간 동안 이 약 40mg을 격주로 피하투여 받았다.
그 결과 이 약을 투여 받은 환자는 위약을 투여 받은 환자와 비교하여 강직성 척추염의 징후 및 증상이 통계적으로 유의하게 개선되었다. 그림 4와 표 11 및 12에 제시한 바와 같이, 질병 활성도 측정으로 확인한 유의한 개선은 2주차에 처음 관찰되었고 24주까지 유지되었다.
전체 척추 강직증이 있는 환자는 이 시험에 포함되었다(n=11). 이 환자들의 반응은 전체 강직증이 없는 환자들과 유사하였다.
표 11: 위약대조 AS 시험에서의 유효성 결과 ‑ AS 시험 I 징후 및 증상 감소
***,** 2, 12, 24주차에서 이 약과 위약 사이의 모든 비교에 대해 p0.001, p0.01에서 통계적으로 유의함
a 강직성 척추염에서의 평가
b 강직성 척추염 활동 지수
위약을 투여 받은 환자 6%와 비교하여 이 약을 투여 받은 환자 22%가 24주차에 낮은 수준의 질병 활성도(4개의 ASAS 반응 매개변수 각각의 수치가 20 [0 ‑100mm 척도])에 도달하였다 (p0.001).
표 12: 강직성 척추염 질병 활성도의 구성요소
a 시각적 아날로그 척도(VAS) 0점 = “없음” 및 100점 = “중증”으로 측정 시에, 최소 20% 및 10점이 개선된 대상자의 비율
b BASDAI(‘d’에서 정의) 5번 및 6번 질문의 평균
c 강직성 척추염 기능 지수
d 강직성 척추염 질병 활성도 지수
e 강직성 척추염 운동능력 지수
f C ‑반응성 단백질(mg/dL)
* 24주차에 이 약과 위약 사이의 모든 비교에 대해 통계적으로 유의함
2건의 시험 중 규모가 작은 무작위, 이중눈가림, 위약대조 시험(AS 시험 II, M03 ‑606)의 강직성 척추염 환자 82명에서도 비슷한 경향이 확인되었다.
환자 보고 결과는 2건의 강직성 척추염 시험 모두에서 일반적인 건강 상태 설문조사 SF ‑36 및 질병 특이적 강직성 척추염 삶의 질 설문조사(Ankylosing Spondylitis Quality of Life Questionnaire; ASQoL)를 사용하여 평가하였다. 12주차의 SF ‑36 신체적 구성요소 점수는 이 약을 투여 받은 환자(평균 변화 6.93)가 위약을 투여 받은 환자(평균 변화 1.55, p0.001)에 비해 유의하게 개선되었으며 이는 24주차까지 유지되었다(평균 변화 7.44 vs. 1.85).
ASQoL의 결과는 전반적인 삶의 질 개선을 입증하는 이 결과를 뒷받침한다. 이 약을 투여 받은 환자(평균 변화 ‑3.15, p0.001)는 위약을 투여 받은 환자(평균 변화 ‑0.95, p0.001)와 비교하여 12주차에 통계적으로 유의하게 개선되었으며, 이는 24주차까지 유지되었다(평균 변화 ‑3.58 vs ‑1.06).
비방사선학적 축성 척추관절염(방사선학적으로 강직성 척추염이 확인되지 않는 축성 척추관절염)
비방사선학적 축성 척추관절염(non ‑radiographic axial spondyloarthritis; nr ‑asSpA) 환자를 대상으로 2건의 무작위, 이중눈가림, 위약대조 시험에서 이 약의 안전성 및 유효성을 평가하였다. nr ‑axSpA I 시험에서는 활동성 nr ‑axSpA가 있는 환자를 평가하였다. nr ‑axSpA II 시험은 공개 연장 이 약 치료 동안 질병 관해에 도달한 활동성 nr ‑axSpA 환자를 대상으로 한 치료 중단 시험이었다.
nr ‑axSpA I 시험
무작위배정, 12주 이중눈가림, 위약대조 시험인 nr ‑axSpA I 시험에서는 활동성 nr ‑axSpA(질병 활성도의 평균 베이스라인 점수[강직성 척추염 질병 활성도 지수(BASDAI)]가 이 약 투여군의 경우 6.4, 위약군의 경우 6.5) 환자 185명을 대상으로 이 약 40 mg 격주 투여를 평가하였으며, 이 환자들은 ≧1개의 NSAID에 대해 부적절한 반응 또는 불내성이 있거나 NSAID가 금지된 환자였다. 여기에 포함된 환자들은 ASAS 축성 SpA 기준에 따라 분류되었고, 강직성 척추염에 대해 수정된 뉴욕 기준을 충족하는 환자 및 건선이나 건선성 관절염이 있는 환자는 제외하였다. 일차 유효성 평가변수는 12주차에 ASAS 40 반응에 도달한 환자 비율이었다.
베이스라인에서 33명(18%)의 환자는 질병 조절 항류마티스제를 병용 투여 받았고 146명(79%)은 NSAID를 병용 투여 받았다. 이중눈가림 기간 후 공개 연장 기간 동안 환자는 최대 144주간 추가적으로 이 약 40 mg을 격주 피하투여 받았다. 12주차 결과에 따르면 전체 치료군 및 양성 MRI 또는 증가된 CRP를 보인 환자 모두에서 위약과 비교하여 이 약을 투여 받은 환자의 활동성 nr ‑axSpA 징후 및 증상이 통계적으로 유의하게 개선되었다(표 13). nr ‑axSpA의 징후 및 증상이 감소했음을 입증한 변수는 24주차 및 68주차에서도 지속되거나 계속 개선되었으며 156주차까지 유지되었다.
표 13: 위약대조 시험 nr ‑axSpA I에서의 유효성 결과#
염증의 억제
이 약을 투여 받은 환자에서 hs ‑CRP와, 천장관절 및 척추의 MRI로 측정한 염증 징후의 유의한 개선은 각각 156주차와 104주차까지 유지되었다. 천장관절에 대한 SPARCC MRI는 131명의 환자에서 이용 가능했고 척추에 대한 SPARCC MRI는 130명의 환자에서 이용 가능했으며 베이스라인으로부터 104주차까지의 평균 변화는 각각 ‑3.8 및 ‑1.4였다.
삶의 질 및 신체적 기능
건강 관련 삶의 질 및 신체적 기능은 HAQ ‑S 및 SF ‑36 설문조사를 사용하여 평가하였다. 이 약은 위약과 비교하여 베이스라인으로부터 12주차까지 HAQ ‑S 전체 점수 및 SF ‑36 신체적 구성요소 요약(Physical Component Summary; PCS)에 대해 통계적으로 유의하게 큰 개선을 보였다. SF ‑36 PCS 점수 및 HAQ ‑S 전체 점수 결과는 각각 52주차, 68주차 및 156주차까지 지속되었다.
nr ‑axSpA II 시험
≧2개의 NSAID에 대해 부적절한 반응을 보이거나 NSAID에 불내성 또는 금지 사유가 있는 활동성 nr ‑axSpA(평균 베이스라인 질병 활성도[BASDAI]가 7.0) 환자 673명은 nr ‑axSpA II 공개 연장 시험의 표지 기간에 등록되어 28주간 격주로 이 약 40mg을 투여 받았다. 이 환자들은 천장관절이나 척추의 염증에 대해 MRI 또는 증가된 hs ‑CRP와 같은 객관적 증거를 가지고 있었다. 공개 연장 시험 기간 동안 최소 12주차에 지속 관해에 도달한 환자(N=305)(16, 20, 24 및 28주차 ASDAS 1.3)는 이중눈가림, 위약대조 기간(총 시험 기간 68주)의 추가 40주 동안 이 약 40mg 격주 투여군(N=152) 또는 위약군(N=153)으로 무작위 배정되었다. 이중눈가림 기간 동안 flared대상자는 최소 12주 동안의 이 약 40mg 격주 구조요법이 허용되었다.
일차 유효성 평가변수는 시험의 68주차까지 재발이 없는 환자 비율이었다. 재발은 4주 간격의 2회 연속 방문에서 ASDAS ≧ 2.1로 정의하였다. 이 약을 투여 받은 대다수의 환자는 위약을 투여 받은 환자와 비교하여 이중눈가림 기간 동안 질병 재발이 없었다(70.4% vs. 47.1%, p0.001)(그림 5).
그림 5: nr ‑axSpA II에서 재발까지의 시간을 요약한 카플란 ‑메이어 곡선
치료 중단에 배정된 그룹에서 재발한 환자 68명 중 65명은 12주간의 이 약 구조요법을 완료하였고 이 중 37명(56.9%)은 공개 연장 시험을 재개하고 12주 후에 다시 질병 관해에 도달하였다(ASDAS 1.3).
68주까지 지속적으로 이 약을 투여 받은 환자는 이중눈가림 기간 동안 치료 중단으로 배정된 환자와 비교하여 활동성 nr ‑axSpA의 징후 및 증상에 대해 통계적으로 유의하게 큰 개선을 보였다(표 14).
표 14: nr ‑axSpA II 위약대조 기간에서의 유효성 결과
크론병
무작위배정, 이중눈가림, 위약대조 시험에서 중등증부터 중증의 활동성 크론병(크론병 활성도 지수[Crohn’s Disease Activity Index; CDAI]가 ≧220 및 ≦450)이 있는 성인 환자를 대상으로 이 약 다중 투여의 안전성과 유효성을 평가하였다. 안정적 용량의 아미노살리실산염, 코르티코스테로이드 및/또는 면역조절제의 투여가 허용되었으며 79%의 환자는 이러한 약물 중 최소 하나의 투여를 지속하였다.
2건의 시험에서 임상적 관해(CDAI 150의 정의)의 유도를 평가하였다. CD 시험 I (M02 ‑403)에서 TNF 차단제 투여 이력이 없는 환자 299명은 다음의 치료군 중 하나로 무작위 배정되었다: 0주차와 2주차에 위약을 투여 받는 위약군, 0주차에 이 약 160 mg과 2주차에 이 약 80 mg을 투여 받는 160/80 투여군, 0주차에 이 약 80 mg과 2주차에 이 약 40 mg을 투여 받는 80/40 투여군, 0주차에 이 약 40mg과 2주차에 이 약20 mg을 투여 받는 40/20 투여군. 임상 결과는 4주차에 평가하였다.
두 번째 유도 시험인 CD 시험 II (M04 ‑691)에서, 인플릭시맙 치료에 반응을 보이지 않거나 불내성이 있는 325명의 환자는 0주차에 이 약 160 mg과 2주차에 이 약 80 mg을 투여 받거나 0주차와 2주차에 위약을 투여 받는 집단으로 무작위 배정되었다. 임상 결과는 4주차에 평가하였다.
CD 시험 III (M02 ‑404)에서 임상적 관해의 유지를 평가하였다. 이 시험에서 활동성 질병이 있는 환자 854명은 공개라벨 이 약을 0주차에 80 mg, 2주차에 40 mg 투여 받았다. 그런 다음 환자는 4주차에 격주 40 mg 이 약 투여군, 매주 40 mg 이 약 투여군, 또는 위약군으로 무작위 배정되었다. 전체 시험 기간은 56주였다. 4주차에 임상 반응을 보인 환자(CR ‑70 = CDAI ≧70 감소)는 4주차에 임상 반응을 보이지 않은 환자와 별도로 계층화하여 분석하였다.
임상적 관해의 유도
TNF 차단제 투여 이력이 없거나(CD 시험 I), 또는 인플릭시맙 치료에 반응을 보이지 않거나 불내성이 있는 것(CD 시험 II)과는 상관 없이 160/80 mg 이 약을 투여 받은 환자의 대부분은 4주차에 위약과 비교하여 임상적 관해를 유도하였다(표 15 참조).
표 15: CD 시험 I 및 CD 시험 II에서 임상적 관해의 유도(환자 비율)
임상적 관해의 유지
CD 시험 III의 4주차에 환자의 58%(499명/854명)가 임상 반응을 보였고 일차 분석에서 평가되었다. 4주차에서 임상 반응을 보인 환자의 대부분이 26주차와 56주차의 위약 유지군 환자와 비교하여 이 약 40 mg 격주 유지군에서 임상적 관해에 도달하였다(표 16 참조). 매주 이 약 치료를 받은 집단은 이 약을 격주로 투여 받은 집단과 비교하여 유의하게 높은 관해율을 입증하지 못했다.
표 16: CD 시험 III에서 임상적 관해의 유지(환자 비율)
임상적 관해는 CDAI 점수 150으로 정의한다. 임상적 반응은 최소 70점의 CDAI 감소로 정의한다.
* 비율에 대한 이 약 vs. 위약 쌍비교, p0.001
시험 동안 관해에 도달한 4주차 반응을 보인 환자 중 격주로 이 약을 투여 받은 환자가 위약 유지군의 환자보다 더 장기간 관해를 유지하였다. 56주차에 질병과 관련된 입원 및 수술은 위약과 비교하여 이 약을 투여 받은 환자에서 통계적으로 유의하게 감소하였다.
CD 시험 I의 환자 117명/276명과 CD 시험 II 및 III의 환자 272명/777명은 최소 3년의 공개라벨 이 약 치료 동안 추적되었다. 각각 88명(75.2%)과 189명(69.5%)의 환자는 임상적 관해가 지속되었다. 임상 반응(CR ‑70)은 각각 107명(91.5%)과 248명(91.2%)의 환자에서 유지되었다.
117명/854명(CD 시험 III)은 스크리닝과 베이스라인 시점 모두에서 배출 누공(draining fistula)을 가지고 있었다. 누공 치유의 평가를 위해 시험에서 사용한 아달리무맙의 두 투여량 모두에 대한 데이터를 통합하였다. 26주차에 누공 치유된 환자의 비율은 아달리무맙을 투여 받은 환자(21명/70명[30.0%])가 위약을 투여 받은 환자(6명/47명[12.8%])에 비해 통계적으로 유의하게 높았다. 완전한 누공 치유는 아달리무맙 투여군과 위약 투여군의 각각 23명/70명(32.9%)과 6명/47명(12.8%)에서 56주차까지 유지되었다.
135명의 환자가 등록된 내시경 시험(M05 ‑769)에서 점막 치유에 대한 이 약의 영향을 확인하였다. 12주차의 점막 치유는 이 약 투여군 중 27.4%, 위약 투여군 중 13.1%에서 확인되었고(p=0.056) 52주차의 점막 치유는 이 약을 투여 받은 환자 중 24.2%, 위약을 투여 받은 환자 중 0%에서 확인되었다(p0.001).
삶의 질
CD 시험 I과 CD 시험 II에서 이 약 80/40 mg 및 160/80 mg 투여군으로 무작위 배정된 환자는 위약군 환자에 비해 질병 특이적 염증성 장질환 설문조사(inflammatory bowel disease questionnaire; IBDQ) 전체 점수에서 통계적으로 유의한 개선을 보였으며, CD 시험 III의 경우 26주차와 56주차에 위약군 환자와 비교하여 아달리무맙 치료군 사이에서도 이러한 개선이 관찰되었다.
궤양성 대장염
2건의 무작위, 이중눈가림, 위약대조 시험에서 중등증부터 중증의 활동성 궤양성 대장염(Mayo 점수 6 ‑12점, 내시경 항목 점수 2 ‑3점)이 있는 성인 환자를 대상으로 이 약 다중 투여의 안전성과 유효성을 평가하였다. 안정적 용량의 아미노살리실산염, 코르티코스테로이드 및/또는 면역조절제의 병용 투여가 허용되었다.
UC ‑I 시험에서 임상적 관해의 유도(Mayo ≦2점, >1점인 항목별 점수는 없는 것으로 정의)를 평가하였다. UC ‑I 시험에서 TNF 길항제 투여 이력이 없는 환자 390명은 0주차와 2주차에 위약 투여군, 0주차에 160 mg 이 약투여 후 2주차에 80 mg 투여군, 또는 0주차에 80 mg 이 약 투여 후 2주차에 40 mg 투여군으로 무작위 배정되었다. 2주차 후에 두 아달리무맙 투여군의 환자는 아달리무맙을 격주로 40 mg 투여 받았다. 임상적 관해는 8주차에 평가되었다.
UC ‑II 시험에서 248명의 환자는 0주차에 160 mg 이 약, 2주차에 80 mg 및 이후 격주로 40 mg 투여 받았으며 246명의 환자는 위약을 투여 받았다. 임상 결과는 8주차에 관해의 유도, 52주차에 관해의 유지에 대해 평가하였다.
160/80 mg 이 약을 투여 받은 대상자는 위약과 비교하여 UC ‑I 시험(각각 18% vs. 9%, p=0.031)과 UC ‑II 시험(각각 17% vs. 9%, p=0.019)에서 통계적으로 유의하게 큰 비율로 8주차에 임상적 관해에 도달하였다. UC ‑II 시험의 8주차에 관해에 도달했던 이 약 투여군 중 21명/41명(51%)은 52주차에 관해를 유지하였다.
전체 UC ‑II 시험군과 전체 Mayo 점수당 치료 8주차에 반응한 환자의 결과는 표 17에 제시한다.
표 17: UC ‑II 시험에서 반응, 관해 및 점막 치유(환자 비율)
UC I와 UC II 시험의 통합 분석에서 입원의 모든 원인 및 UC와 관련된 입원율이 통계적으로 유의하게 감소했음을 관찰하였다.
UC ‑II 시험에서 약 40%의 환자가 인플릭시맙을 통한 항 ‑TNF 치료에 실패한 이력이 있었다. 이러한 환자에서 아달리무맙의 효능은 항 ‑TNF 투여 이력이 없는 환자와 비교하여 감소하였다. 항 ‑TNF 치료에 실패 이력이 있는 환자 중 52주차 관해율은 위약군에서 3%, 아달리무맙군에서 10%에 도달하였다.
UC 시험 I 및 II의 환자는 공개라벨 장기간 확장 시험(UC ‑III)으로 전환할 수 있는 선택권이 있었다. 3년간의 이 약 치료 후 74%(268명/360명)의 환자에서 부분 Mayo 점수당 임상적 관해가 지속되었고 최소 4년간 이 약 치료를 받은 환자의 경우 75%(97명/130명)가 부분 Mayo 점수당 임상적 관해가 지속되었다. 1년 이상의 치료 후 반응 도달에 실패한 환자는 매주 40 mg으로 투여 빈도를 증가시킴으로써 치료 이익을 받을 수 있었다.
삶의 질
UC 시험 II에서 이 약 160/80 mg 투여군으로 무작위 배정된 환자는 위약과 비교하여 52주차에 질병 특이적 염증성 장질환 설문조사(IBDQ) 전체 점수의 개선에 도달하였다(p=0.007).
베체트 장염
기존의 치료(스테로이드 또는 면역억제제)에 부적절한 반응을 보인 장내 베체트병이 있는 일본인 환자 대상의 공개, 비통제(uncontrolled) 시험에서 24주차에 현저한 개선율은 45.0%(9명/20명)였다. 현저한 개선은, 종합적인 질병 평가 척도를 사용하여 위장 증상의 전반적 평가와 내시경 평가 점수 모두 ≦1점인 환자의 비율로 정의하였고 이는 궤양의 크기 및 위장 증상 개선을 둘 다 반영한다.
판상 건선
무작위배정, 이중눈가림 시험에서 전신 치료 또는 광선치료의 대상자인 만성 판상 건선( ≧10% BSA 관련, 건선 영역 및 중증도 지수[Psoriasis Area and Severity Index; PASI] ≧12 또는 ≧10)이 있는 성인 환자에서 이 약의 안전성과 유효성을 시험하였다. 건선 시험 I과 II에 등록된 환자 중 73%는 전신 치료나 광선치료를 받은 이력이 있었다. 또한 무작위 배정된 이중눈가림 시험(건선 시험 III)에서 전신 치료의 대상자인 손 및/또는 발의 건선을 동반한 중등증부터 중증의 만성 판상형 건선이 있는 성인 환자에서도 이 약의 안전성과 유효성을 시험하였다.
건선 시험 I (M03 ‑656)은 3개의 치료 기간 동안 1,212명의 환자를 평가하였다. A 기간에 환자는 위약을 투여 받거나 초기 용량 80 mg의 이 약을 투여 받은 후 일주일 뒤부터 격주로 40 mg을 투여 받았다. 16주간의 치료 후 최소 PASI 75 반응(베이스라인과 비교하여 최소 75%의 PASI 점수 개선)에 도달한 환자는 B 기간에 진입하여 공개라벨 40 mg 이 약을 격주로 투여 받았다. 33주차에 ≧PASI 75 반응이 유지되고 기존에 A 기간에서 활성 치료로 무작위 배정된 환자는 C 기간에 무작위 재배정되어 추가 19주 동안 격주로 40 mg 이 약 또는 위약을 투여 받았다. 모든 치료군에서 평균 베이스라인 PASI 점수는 18.9점이었고 베이스라인에서 의사의 전반적 평가(Physician’s Global Assessment; PGA) 점수의 범위는 “중등증”(환자의 53%) ‑“중증”(41%) ‑“매우 중증”(6%)이었다.
건선 시험 II (M04 ‑716)에서는 271명의 환자에서 메토트렉세이트 및 위약에 대해 이 약의 안전성과 유효성을 비교하였다. 환자는 위약, 메토트렉세이트 초기 용량인 7.5mg을 투여 받은 후 12주차까지 최대 25 mg으로 증량하여 투여 받거나, 이 약 초기 용량 80 mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 40 mg을 투여 받았다. 16주 이상의 치료를 통해 이 약과 메토트렉세이트를 비교한 이용가능한 데이터는 없다. MTX를 투여 받고 8주차 및/또는 12주차에 ≧PASI 50 반응에 도달한 환자는 추가로 투여량을 증량하지 않았다. 모든 치료군에서 평균 베이스라인 PASI 점수는 19.7점이었고 베이스라인 PGA 점수의 범위는 “경증”(1%) ‑“중등증”(48%) ‑“중증”(46%) ‑“매우 중증”(6%)이었다.
임상2상 및 임상3상 건선 시험에 모두 참여한 환자는 이 약을 추가로 최소 108주 동안 투여 받는 공개라벨 확장 시험(M03 ‑658)에 등록할 수 있었다.
건선 시험 I과 II에서 일차 평가변수는 베이스라인으로부터 16주차에 PASI 75 반응에 도달한 환자의 비율이었다(표 18 및 19 참조).
표 18: 16주차의 Ps 시험 I (REVEAL) 유효성 결과
표 19: 16주차의 Ps 시험 II (CHAMPION) 유효성 결과
건선 시험 I에서 PASI 75 반응에 도달하여 33주차에 위약으로 무작위 재배정된 환자의 28%는 이 약을 지속적으로 투여 받은 환자의 5%와 비교하여(p0.001) “적절한 반응의 실패(loss)”을 경험하였다(베이스라인과 비교하여 PASI 50 반응을 나타내고 33주차와 비교하여 PASI 점수가 6점 이상 감소한, 33주 후 52주차 이하의 PASI 점수). 위약으로 무작위 재배정된 후 적절한 반응 도달에 실패하여 공개라벨 확장 시험에 등록된 환자 중 38% (25명/66명)과 55% (36명/66명)는 각각 12주와 24주의 재치료를 받은 후 PASI 75 반응에 다시 도달하였다.
16주차와 33주차의 PASI 75 반응자 총 233명은 건선 시험 I에서 52주 동안 지속적으로 이 약을 투여 받았으며 공개라벨 확장 시험에서 이 약을 지속적으로 투여 받았다. 이 환자들에서 PASI 75와 PGA 개선 또는 최소 반응률은 공개라벨에서의 추가 108주 치료 후 각각 74.7%와 59.0%였다(총 160주).
건선 시험 II에서 이 약 치료로 무작위 배정된 총 94명은 공개라벨 확장 시험에서 이 약을 지속적으로 투여 받았다. 이 환자들에서 PASI 75와 PGA 개선 또는 최소 반응률은 공개라벨에서의 추가 108주 치료 후 각각 58.1%와 46.2%였다(총 124주).
총 347명의 안정적인 반응자가 공개라벨 확장 시험에서 중단 및 재치료 평가에 참여하였다. 재발까지의 시간 중앙값(PGA가 “중등증” 또는 최악으로 감소)은 약 5개월이었다. 중단 기간 동안 반동현상(rebound)을 경험한 환자는 없었다. 재치료 기간에 등록된 환자 중 76.5% (218명/285명)은 중단 동안의 재발 여부와 관계 없이 재치료 16주 후 PGA “개선” 또는 “최소” 반응을 보였다(중단 기간 동안 재발 또는 재발되지 않은 환자에서 각각 69.1% [123명/178명], 88.8% [95명/107명]).
위약(시험 I 및 II) 및 MTX(시험 II)와 비교하여 베이스라인으로부터 16주차에 DLQI(피부학적 삶의 질 지수)에서 유의한 개선이 입증되었다. 시험 I에서 SF ‑36의 신체적 및 정신적 구성요소 요약 점수 또한 위약과 비교하여 유의하게 개선되었다.
공개라벨 확장 시험에서 PASI 반응이 50% 미만으로 감소하여 격주 40mg에서 매주 40mg으로 투여량을 증량한 환자 중 각각 26.4% (92명/349명)와 37.8% (132명/349명)는 12주차와 24주차에 PASI 75 반응에 도달하였다.
건선 시험 III (REACH)은 중등증부터 중증의 만성 판상형 건선과 함께 손 및/또는 발 건선이 있는 72명의 환자를 대상으로 위약에 대해 이 약의 효능 및 안전성을 비교하였다. 환자는 이 약 초기 용량 80mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 이 약 40mg을 투여 받거나 위약을 투여 받았다. 16주차에 통계적으로 유의하게 많은 비율의 이 약 투여군 환자가 위약군 환자에 비해 손 및/또는 발에 대해 PGA ‘개선’ 또는 ‘거의 개선’에 도달하였다(각각 30.6% vs. 4.3% [P=0.014]).
건선 시험 IV는 중등증부터 중증의 손발톱 건선이 있는 성인 환자 217명에서 위약에 대해 이 약의 안전성과 유효성을 비교하였다. 환자는 이 약 초기 용량 80mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 이 약 40mg을 투여 받거나 위약을 투여 받았고, 추가 26주 동안 공개라벨 이 약 치료를 받았다. 손발톱 건선은 수정된 손발톱 건선 중증도 지수(mNAPSI) 및 손톱 건선에 대한 의사의 전반적 평가(PGA ‑F)를 사용하여 평가하였다. 위약에 무작위 배정된 환자와 비교하여 이 약에 무작위 배정된 환자 중 통계적으로 유의하게 높은 비율의 환자가 26주차에 mNAPSI에서 최소 75% 개선(mNAPSI 75)에 도달하였다(표 20 참조). NAPSI의 개선율은 이 약 투여군 환자가 위약군 환자보다 16주차(44.2% vs.7.8%)와 26주차(56.2% vs 11.5%)에 통계적으로 유의하게 높았다.
이 약 투여군에서 통계적으로 유의하게 높은 비율의 환자는 위약군과 비교하여 26주차에 베이스라인으로부터 2등급 이상 개선하여 PGA ‑F “개선” 또는 “최소”에 도달하였다. 이 시험에서 이 약은 다양한 범위(BSA ≧10%와 BSA 10% 및 ≧5%)의 손발톱 건선 환자에서 치료 이익을 입증하였으며 위약과 비교하여 두피 건선에서도 통계적으로 유의한 개선을 보였다.
표 20: 26주차의 유효성 결과
52주차까지 이 약 치료를 지속적으로 투여 받은 환자 중 65.0%가 mNAPSI 75 반응에 도달하였고 61.3%는 PGA ‑F 반응에 도달하였다.
이 약을 투여 받은 환자는 위약과 비교하여 26주차의 DLQI(피부학적 삶의 질 지수)가 베이스라인으로부터 통계적으로 유의하게 개선되었다. 26주차에 베이스라인으로부터의 평균 감소(개선)는 이 약군(N=94)에서 8.0, 위약군(N=93)에서 1.9였다.
화농성 한선염
전신 항생제 치료에 불내성 또는 부적절한 반응을 보이거나 금지 사유가 있는 중등증에서 중증의 화농성 한선염(hidradenitis suppurativa; HS) 성인 환자를 대상으로 무작위, 이중눈가림, 위약대조 시험에서 이 약의 안전성과 유효성을 평가하였다. HS ‑I 시험과 HS ‑II 시험의 환자는 최소 3개의 농양 또는 염증성 결절이 있는 Hurley II단계 또는 III단계 질병을 가지고 있었다.
HS ‑I 시험(M11 ‑313)은 2개의 치료 기간에서 307명의 환자를 평가하였다. A 기간에서 환자는 0주차에 위약 또는 이 약 초기 용량 160 mg, 2주차에 80 mg, 4주차부터 11주차까지 매주 40mg을 투여 받았다. 항생제 병용 투여는 시험 동안 허용되지 않았다. 12주간의 치료 후 A 기간에 이 약을 투여 받은 환자는 B 기간에 3개의 치료군 중 하나로 무작위 재배정되었다(12주차부터 35주차까지 매주 이 약 40mg 투여군, 격주 이 약 40mg 투여군, 또는 위약군). A 기간에 위약으로 무작위 배정된 환자는 B 기간에 매주 이 약 40mg 투여군으로 배정하였다.
HS ‑II 시험(M11 ‑810)은 2개의 치료 기간에서 326명의 환자를 평가하였다. A 기간에서 환자는 0주차에 위약 또는 이 약 초기 용량 160 mg, 2주차에 80 mg, 4주차부터 11주차까지 매주 40 mg을 투여 받았다. 환자 중 19.3%는 시험 동안 베이스라인의 경구 항생제 치료를 지속하였다. 12주간의 치료 후 A 기간에 이 약을 투여 받은 환자는 B 기간에 3개의 치료군 중 하나로 무작위 재배정되었다(12주차부터 35주차까지 매주 이 약 40 mg 투여군, 격주 이 약 40 mg 투여군, 또는 위약군). A 기간에 위약으로 무작위 배정된 환자는 B 기간에 위약군으로 배정하였다.
HS ‑I 시험과 HS ‑II 시험에 참여한 환자는 이 약 40 mg을 매주 투여 받는 공개라벨 확장 시험에 등록될 수 있었다. 모든 아달리무맙 투여군의 평균 노출은 762일이었다. 3개의 치료군 모두 국소 소독 세척을 매일 실행하였다.
임상 반응
염증 병변의 임상 반응은 화농성 한선염 임상 반응(HiSCR: 전체 농양 및 염증성 결절 수가 최소 50% 감소하고 베이스라인과 비교하여 농양 수가 증가하거나 배출 누공의 수가 증가하지 않음)을 사용하여 평가하였다. HS와 관련된 피부 통증의 감소는 11점 척도에서 초기 베이스라인 점수가 3점 이상이었던 환자에서 수치 등급 척도를 사용하여 평가하였다.
12주차에 위약과 비교하여 유의하게 높은 비율의 이 약 투여 환자가 HiSCR에 도달하였다. 12주차에 HS ‑II 시험에서 유의하게 높은 비율의 환자는 HS와 관련된 피부 통증이 임상적으로 의미 있게 감소됨을 경험하였다(표 21 참조). 이 약을 투여 받은 환자는 치료 초기 12주 동안 질병 재발의 위험이 유의하게 감소하였다.
표 21: HS 시험 I과 II의 12주차에서의 유효성 결과
지속적으로 이 약을 매주 투여 받는 집단에 무작위 배정된 환자의 12주차 전체 HiSCR 비율은 96주차까지 유지되었다. 96주 동안 매주 이 약 40 mg을 장기간 투여 받은 환자에서 새로운 안전성 결과는 확인되지 않았다.
위약과 비교하여 베이스라인으로부터 12주차의 큰 개선은 피부학적 삶의 질 지수(DLQI)로 측정한 피부 특이적 건강 관련 삶의 질(HS ‑I 시험 및 HS ‑II 시험), 치료 만족 설문조사 ‑ 약물(TSQM)로 측정한 환자의 전반적인 약물 치료 만족(HS ‑I 시험 및 HS ‑II 시험), 그리고 SF ‑36의 신체적 구성요소 요약 점수로 측정한 신체적 건강(HS ‑I 시험)으로 입증하였다.
포도막염
독립적인 앞포도막염 환자를 제외하고 비감염성 중간포도막염, 후포도막염 및 전체포도막염(“후부에 영향을 미치는 비감염성 포도막염”으로도 알려짐)이 있는 성인 환자를 대상으로 2건의 무작위, 이중눈가림, 위약대조 시험(UV 시험 I [M10 ‑877] 및 UV 시험 II [M10 ‑880])에서 이 약의 안전성과 유효성을 평가하였다. 환자는 위약을 투여 받거나 초기 용량 80mg의 이 약을 투여 받은 후 일주일 뒤부터 격주로 40mg을 투여 받았다. 안정적 용량의 비생물학적 면역억제제를 병용 투여하는 것은 허용되었다. 두 시험의 일차 효능 평가변수는 ‘치료 실패까지의 시간’이었다. 초기 질병 제어 이후 치료 실패까지의 시간을 연장하면 질병 재발, 염증 및 시력 상실의 위험이 감소된다.
치료 실패는 염증성 맥락망막 및/또는 염증성 망막 혈관 병변, 전방(anterior chamber; AC) 세포 등급, 유리체 혼탁(vitreous haze; VH) 등급 및 최대교정시력(best corrected visual acuity; BCVA)를 기반으로 복합적인 구성요소 결과를 통해 정의하였다.
UV 시험 I은 코르티코스테로이드(10 ‑60mg/일의 경구 프레드니손) 치료에도 불구하고 활동성 포도막염이 있는 환자 217명을 평가하였다. 모든 환자는 시험 등록시 프레드니손의 표준 용량 60mg/일을 투여 받은 후 의무적으로 테이퍼링(tapered) 계획에 따라 코르티코스테로이드를 15주차까지 중단하였다.
UV 시험 II는 베이스라인에서 질병 제어를 위해 만성 코르티코스테로이드(10 ‑35 mg/일의 경구 프레드니손) 치료가 필요한 비활동성 포도막염 환자 226명을 평가하였다. 환자는 이후 의무적으로 테이퍼링 계획에 따라 코르티코스테로이드를 19주차까지 중단하였다.
임상 반응
두 시험 결과는 이 약을 투여 받은 환자가 위약을 투여 받은 환자와 비교하여 치료 실패의 위험이 통계적으로 유의하게 감소했음을 입증하였다(표 22 참조). 두 시험은 위약과 비교하여 치료 실패율에 대한 이 약의 초기에 지속적인 영향을 입증하였다(그림 6 참조).
표 22: UV 시험 I과 II의 치료 실패까지의 시간
그림 6: 6주차 이후(UV 시험 I) 또는 2주차 이후(UV 시험 II)
치료 실패까지의 시간을 요약한 카플란 ‑메이어 곡선
두 시험 모두에서 일차 평가변수의 모든 요소는 이 약군과 위약군 사이의 전반적인 차이에 점증적으로 기여하였다(표 23).
표 23: UV 시험 I과 II의 치료 실패 구성 요소
또한 UV 시험 I에서 AC 세포 등급, 유리체 혼탁 등급 및 logMAR BCVA(6주차 전의 최적 상태에서 마지막 방문까지의 평균 변화, P값은 각각 0.011, 0.001 및 0.003)의 변화에 대해 위약과 비교하여 아달리무맙에 유리하게 통계적으로 유의한 차이가 관찰되었다.
UV 시험 I 및 UV 시험 II의 비통제 장기간 확장에 포함된 417명의 환자 중 46명이 부적합한 것으로 간주되었고(예: 당뇨망막병증에 의한 합병증 진행, 백내장 수술 또는 유리체절제술로 인해) 효능의 일차 분석에서 제외되었다. 나머지 371명의 환자 중 276명의 평가 가능한 환자는 78주간의 공개라벨 아달리무맙 치료를 완료하였다. 관찰된 데이터 접근법을 기반으로, 222명(80.4%)은 매일 ≦7.5mg의 스테로이드 투여를 동반한 무활동기였고(활동성 염증성 병변 없음, AC 세포 등급 ≦0.5+, VH 등급 ≦0.5+) 184명(66.7%)은 스테로이드를 투여하지 않는 무활동기였다. BCVA는 78주차에 눈의 88.4%에서 개선 또는 유지되었다(5문자 미만으로 하락). 78주 전에 시험을 중단한 환자 중 11%는 이상사례, 5%는 아달리무맙 치료에 대한 불충분한 반응으로 인해 중단하였다.
삶의 질
UV 시험 I에서 이 약 치료는 NEI VFQ ‑25로 측정한 시력과 관련된 기능 및 건강 관련 삶의 질을 유지시켰다.
⓶ 소아
소아 특발성 관절염
다관절성 소아 특발성 관절염(pJIA)
다양한 소아 특발성 관절염 발생 유형(가장 흔한 류마티스인자 음성 또는 양성 다관절성 및 확장성 소수 관절염)을 가진 활동성 다관절성 또는 다관절 과정 소아 특발성 관절염이 있는 소아를 대상으로 2건의 시험(pJIA I 및 II)에서 이 약의 안전성과 유효성을 평가하였다.
pJIA I
다관절성 소아 특발성 관절염(juvenile idiopathic arthritis; JIA)이 있는 171명의 아동(4 ‑ 17세)을 대상으로 다기관, 무작위, 이중눈가림, 평행군 시험에서 이 약의 안전성과 유효성을 평가하였다. 공개라벨 유도 단계(open ‑label lead in; OL LI)에서 환자는 메토트렉세이트를 투여 받은 환자 또는 메토트렉세이트를 투여 받지 않은 환자의 두 집단으로 계층화되었다. 비 ‑메토트렉세이트층에 있는 환자는 메토트렉세이트 투여 이력이 없거나 시험 약물을 투여하기 최소 2주 전에 MTX를 중단한 환자였다. 환자는 안정적 용량의 NSAID 및/또는 프레드니손( ≦0.2 mg/kg/일 또는 최대 10 mg/일)의 투여를 유지하였다. OL LI 단계에서 모든 환자는 24 mg/m2, 최대 40 mg의 이 약을 16주 동안 격주로 투여 받았다. 연령별 환자 분포 및 OL LI 단계 동안 투여 받은 최소, 중앙값, 최대 용량은 표 24에 제시한다.
표 24: 연령별 환자 분포와 OL LI 단계 동안 투여 받은 이 약 용량
16주차에 소아 ACR 30 반응을 입증한 환자는 이중눈가림(double blind; OB) 단계에서 무작위 배정되어 추가 32주 동안, 또는 질병이 재발할 때까지 이 약 24 mg/m2, 최대 40 mg을 투여 받거나 위약을 격주로 투여 받았다. 질병 재발 기준은 6개의 소아 ACR 핵심 기준 중 3개 이상이 베이스라인으로부터 ≧30% 악화되고, ≧2개의 활동성 관절 및 6개의 기준 중 >30%의 개선을 보이는 기준이 1개를 넘지 않는 것으로 정의하였다. 32주 후 또는 질병 재발 시 환자는 공개라벨 연장 단계에 등록될 수 있었다.
표 25: JIA 시험에서 소아 ACR 30 반응
16주차에 반응을 보인 환자(n=144) 중, 시험 전반에 걸쳐 이 약을 투여 받은 환자는 OLE 단계에서 최대 6년간 ACR 30/50/90 반응이 유지되었다. 전체적으로 19명의 대상자는 6년 이상 이 약을 투여 받았으며 이들 중 11명은 베이스라인 연령군 4 ‑ 12세, 8명은 베이스라인 연령군 13 ‑ 17세였다.
이 약과 메토트렉세이트를 병용 투여했을 경우 이 약을 단독 투여했을 때보다 전반적인 반응은 일반적으로 우수했으며 보다 적게 항체가 발생하였다. 이러한 결과를 고려해볼 때, 이 약을 메토트렉세이트와 병용 투여하는 것이 권장되며 메토트렉세이트의 투여가 적절하지 않은 환자는 단일 요법으로 사용하는 것이 권장된다.
pJIA II
중등증부터 중증의 활동성 다관절성 JIA가 있는 32명의 아동(2세 ‑4세 아동 또는 체중이 15kg인 4세 이상의 아동)을 대상으로 공개라벨, 다기관 시험에서 이 약의 안전성과 유효성을 평가하였다. 환자는 24 mg/m2 체표면적(body surface area; BSA), 최대 20 mg의 이 약을 최소 24주 동안 SC 단일 주입을 통해 격주로 투여 받았다. 시험 동안 대부분의 대상자는 MTX를 병용 투여하고 있었으며 일부에서 코르티코스테로이드 또는 NSAID 병용 투여를 보고하였다.
12주차와 24주차에 관찰된 데이터 접근법을 사용한 소아 ACR 30 반응은 각각 93.5%와 90.0%였다. 12주차와 24주차에 소아 ACR 50/70/90에 도달한 대상자의 비율은 각각 90.3%/61.3%/38.7%와 83.3%/73.3%/36.7%였다. 24주차에 반응(소아 ACR 30)을 보인 환자(30명 중 27명) 중 이 기간 동안 이 약을 투여 받은 환자는 OLE 단계에서 최대 60주 동안 ACR 30 반응이 유지되었다. 전반적으로 20명의 대상자가 60주 이상 이 약을 투여 받았다.
골부착부염 관련 관절염
골부착부염 관련 관절염이 있는 46명의 소아 환자(6 ‑ 17세)를 대상으로 다기관, 무작위, 이중눈가림 시험에서 이 약의 안전성과 유효성을 평가하였다. 환자는 24 mg/m2 체표면적(body surface area; BSA), 최대 40 mg의 이 약 또는 위약을 12주 동안 투여하는 집단 중 하나로 무작위 배정되었다. 이중눈가림 기간 후 공개라벨(open ‑label; OL) 기간 동안 환자는 이 약 24 mg/m2 BSA, 최대 40 mg을 추가 192주 동안 격주로 피하 투여 받았다. 일차 평가변수는 베이스라인부터 12주차까지 관절염(기형 또는 통증 및/또는 압통을 동반한 운동 상실 관절로 인한 것이 아닌 부종)이 있는 활동성 관절 수의 변화율이었으며 이 약군의 평균 비율 감소는 ‑62.6%인 반면 위약군에서는 ‑11.6%였다. 관절염이 있는 활동성 관절 수의 개선은 공개라벨 기간 동안 156주차까지 유지되었다. 환자의 대다수는 부착부염 부위의 수, 압통 관절 수(tender joint count; TJC), 종창 관절 수(swollen joint count; SJC), 소아 ACR 30 반응, 소아 ACR 50 반응 및 소아 ACR 70 반응과 같은 이차 평가변수의 임상적 개선을 입증하였으며, 이러한 개선은 공개라벨 기간 동안 156주차까지 유지되었다.
소아 크론병
소아 크론병 활동도 지수(Pediatric Crohn's Disease Activity Index; PCDAI) 점수가 >30인 것으로 정의되는 중등증부터 중증의 크론병(Crohn’s disease; CD)이 있는 6 ‑ 17세의 소아 환자 192명을 대상으로 체중(40 kg 또는 ≧40 kg)에 따른 이 약의 유도 및 유지 치료에 대한 효능 및 안전성을 평가하기 위해 다기관, 무작위, 이중눈가림 임상시험을 설계하여 이 약을 평가하였다. 대상자는 CD에 대한 기존 치료(코르티코스테로이드 및/또는 면역조절제 포함)에 실패한 이력이 있어야 했다. 또한 대상자는 인플릭시맙에 대한 반응 도달에 실패했거나 불내성이 있을 수 있다.
모든 대상자는 베이스라인 체중을 기반으로 한 투여량으로 공개라벨 유도 치료를 받았다. 체중이 ≧40 kg인 환자의 경우 0주차에 160 mg, 2주차에 80 mg을 투여 받았으며 40 kg인 환자의 경우에는 각각 80 mg과 40 mg을 투여 받았다.
4주차에 대상자는 표 26에 제시된 바와 같이 저용량 또는 표준 용량 유지 요법을 받는 시점의 체중에 따라 각 투여군에 1:1로 무작위 배정되었다.
표 26: 유지 요법
유효성 결과
시험의 일차 평가변수는 26주차에 PCDAI 점수 ≦10점으로 정의되는 임상적 관해였다.
임상적 관해 및 임상 반응(PCDAI 점수가 베이스라인으로부터 최소 15점 감소되는 것으로 정의) 비율은 표 27에 제시한다. 코르티코스테로이드 또는 면역조절제의 중단율은 표 28에 제시한다.
표 27: 소아 CD 시험의 PCDAI 임상적 관해 및 반응
표 28: 소아 CD 시험의 코르티코스테로이드 또는 면역조절제 중단 및 누공 관해
26주차와 52주차에 두 치료군에서 체질량지수 및 신장 속도에 대해 베이스라인으로부터 통계학적으로 유의한 증가(개선)가 관찰되었다. 삶의 질 매개변수(IMPACT III 포함)에 대해서도 두 치료군 모두에서 베이스라인으로부터 통계적 및 임상적으로 유의한 개선이 관찰되었다.
소아 CD 시험의 환자는 공개라벨 장기간 확장 시험에서 투여를 지속할 수 있는 선택권이 있었다. 5년간의 이 약 치료 이후 환자의 74% (37명/50명)은 임상적 관해가 지속되었고 환자의 92% (46명/50명)는 PCDAI에 따라 임상 반응이 지속되었다.
소아 판상 건선
국소 치료 및 일광요법이나 광선요법으로 부적절하게 제어된 중증의 만성 판상형 건선(PGA ≧4 또는 >20% BSA 관련 또는 매우 두꺼운 병변을 동반한 >10 % BSA 관련 또는 PASI ≧20 또는 임상적으로 관련된 얼굴, 생식기, 손/발 병변을 동반한 PASI ≧10으로 정의)이 있는 4세 소아 환자 114명을 대상으로 무작위, 이중눈가림, 통제 시험에서 이 약의 효능을 평가하였다.
환자는 격주 0.8 mg/kg(최대 40mg) 이 약, 격주 0.4 mg/kg(최대 20mg) 이 약, 또는 매주 0.1 ‑0.4 mg/kg(최대 25mg) 메토트렉세이트를 투여 받았다. 16주차에 이 약 0.8 mg/kg 투여군에 무작위 배정된 환자가 MTX에 무작위 배정된 환자보다 더 많은 수에서 호의적인 유효성 반응(예: PASI 75)을 보였다.
표 29: 16주차의 소아 판상 건선 유효성 결과
PASI 75 및 PGA 개선이나 최소에 도달한 환자는 최대 36주간의 치료를 중단하였으며 질병 제어의 실패(PGA 반응 도달 실패)에 대해 모니터링되었다. 그런 다음 환자는 추가 16주 동안 아달리무맙 0.8 mg/kg을 격주로 재투여하였으며 재투여 동안 관찰된 반응은 이전의 이중눈가림 기간에 관찰된 것과 유사하였다: PASI 75 반응 78.9%(19명 중 15명), PGA 개선 또는 최소 52.6 %(19명 중 10명).
시험의 공개라벨 기간에 PASI 75 및 PGA 개선이나 최소 반응은 새로운 안전성 결과 없이 추가적으로 최대 52주 동안 유지되었다.
⓷ 면역원성
항 ‑아달리무맙 항체의 형성은 아달리무맙의 청소율 증가 및 효능 감소와 관련된다. 항 ‑아달리무맙 항체의 존재와 이상반응 사이의 상관관계는 명백하지 않다.
면역원성 분석이 의약품마다 다르기 때문에 다른 의약품의 항체 비율과 비교하는 것은 오해의 소지가 있을 수 있다.
성인
류마티스 관절염 시험 I, II 및 III의 환자에서 6 ‑12개월 동안 다양한 시점에서 항 ‑아달리무맙 항체에 대해 검사하였다. 확증시험에서 항 ‑아달리무맙 항체가 아달리무맙을 투여 받은 환자의 5.5% (58명/1053명)에서 확인되었고, 그에 반해 위약을 투여 받은 환자에서는 0.5% (2명/370명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 12.4%였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 0.6 %였다.
건선성 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 10 % (38명/376명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 13.5 % (24명/178명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 7 % (14명/198명)였다.
강직성 척추염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 8.3 % (17명/204명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 8.6 % (16명/185명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 5.3 % (1명/19명)였다.
비방사선학적 축성 척추관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 지속적으로 투여 받은 환자의 8명/152명(5.3 %)에서 확인되었다.
크론병 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 2.6% (7명/269명)에서 확인되었다.
중등증에서 중증의 활동성 UC 환자에서 아달리무맙을 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 5.0%였다.
장내 베체트병이 있는 일본인 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 5% (1명/20명)에서 확인되었다.
건선 환자에서 항 ‑아달리무맙 항체는 메토트렉세이트를 병용 투여하지 않은 아달리무맙 투여 환자의 8.4 % (77명/920명)에서 확인되었다. 메토트렉세이트를 병용 투여하지 않고 장기간 아달리무맙을 투여하고 중단 및 재투여 시험에 참여한 판상형 건선 환자에서 재투여 후 항 ‑아달리무맙 항체의 발생률은 2.3 %였고 중단 전에 관찰된 발생률인 1.9%와 유사하였다.
중등증에서 중증의 화농성 한선염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 10.1% (10명/99명)에서 확인되었다.
비감염성 포도막염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 4.8% (12명/249명)에서 확인되었다.
소아
4 ‑ 17세의 다관절성 소아 특발성 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 16%에서 확인되었다. 메토트렉세이트를 병용하지 않은 환자의 경우 발생률은 26%였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 6%였다. 2세부터 4세 또는 체중이 15 kg인 4세 이상의 다관절성 소아 특발성 관절염 환자의 7%에서 항 ‑아달리무맙 항체가 확인되었고 1명의 환자는 메토트렉세이트를 병용 투여하는 중이었다.
골부착부위염 관련 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 11% (5명/46명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 14% (3명/22명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 8% (2명/24명)였다.
중등증에서 중증의 활동성 소아 크론병 환자에서 아달리무맙을 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 3.3%였다.
소아 건선 환자에서 0.8 mg/kg 아달리무맙을 단일 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 13% (5명/38명)였다.
3) 비임상 안전성 자료
비임상 데이터에 따르면 아달리무맙은 단회 투여 독성, 반복 투여 독성 및 유전 독성 시험을 근거로 인간에 대해 특별한 위해성을 나타내지 않는다.
배아 태아 발달 독성/주산기 발달 연구는 사이노몰로거스(cynomologous) 원숭이에서 0, 30 및 100 mg / kg (그룹 당 9 ‑17 마리의 원숭이)의 용량으로 수행되었으며, 아달리무맙이 태아에게 해를 입힌다는 증거는 없었다. 설치류 TNF에 대한 교차 반응이 제한적인 항체 및 설치류에서 중화 항체의 개발에 대한 적절한 모델이 없기 때문에 아달리무맙에 대한 발암성 연구나 생식기능 및 출생 후 독성에 대한 표준 평가는 수행되지 않았다.
1) 감염 : 이 약의 투여 전, 투여 동안 및 투여 후 결핵을 포함한 감염에 대해 면밀히 모니터링해야 한다. 이 약의 배설에 5개월까지 소요될 수 있으므로 이 기간 동안 모니터링을 지속해야 한다. 만성 또는 국소화된 감염 등 활동성 감염 환자의 경우 감염이 조절될 때까지 이 약의 투여를 시작해서는 안 된다. 결핵에 노출된 환자 및 결핵의 위험이 높은 지역 또는 히스토플라스마증, 콕시디오이데스진균증, 또는 분아균증(blastomycosis) 같은 풍토병성 진균증의 위험이 높은 지역을 여행한 환자들은, 치료를 시작하기 전 이 약의 치료로 인한 위험 및 이익에 대하여 고려하여야 한다. 이 약의 투여 동안 새로운 감염이 발생한 환자는 면밀히 모니터링해야 하며, 완벽한 진단 평가를 거쳐야 한다. 만약 새로운 심각한 감염 또는 패혈증이 발생한 환자의 경우 감염이 조절될 때 까지 이 약의 투여를 중단해야 하며, 적절한 항균 또는 항진균 치료를 시작해야 한다. 재발성 감염 병력 또는 감염되기 쉬운 기저상태(면역억제제를 병용 투여 하는 경우 포함)에 있는 환자에게 이 약의 투여를 고려하는 의사는 주의를 기울여야 한다.
세균성, 진균성, 침습적 진균(파종성 또는 폐외 히스토플라스마증, 아스페르길루스증, 콕시디오이데스진균증), 바이러스성으로 인한 중대한 감염, 기생충에 의한 감염 또는 기타 기회감염이 TNF 저해제를 투여 받고 있는 환자들에게서 보고되었다. 또한 패혈증, 드물게 결핵, 칸디다증, 리스테리아증, 레지오넬라증 및 뉴모시스티스(pneumocystis)가 이 약을 포함한 TNF 저해제의 사용에서 보고되었다. 감염과 관련된 입원 또는 치명적인 결과가 보고되었으며, 심각한 감염은 대부분 선행 질환과 함께 감염을 일으키기 쉬운 면역억제제를 병용 투여한 환자에서 발생했다.
⓵ 심각한 감염
이 약을 투여받고 있는 환자에게 심각한 감염의 위험이 증가되는 경우가 임상시험에서 나타났으며, 시판 후 조사에서 이러한 사실을 뒷받침하는 보고가 있었다. 특히 폐렴, 신우신염, 패혈성 관절염, 패혈증과 같은 심각한 감염이 보고되었다.
⓶ 결핵
이 약을 투여받고 있는 환자에게서 결핵(재활성화 및 발병 포함)이 보고되었으며, 폐결핵 및 폐외결핵(예, 파종성)이 포함되었다.
이 약으로 치료를 시작하기 전 모든 환자의 활동성 또는 비활동성(잠복성) 결핵 감염에 대해 평가해야 한다. 이 평가에는 개인의 결핵 병력 또는 과거에 활동성 결핵 환자에 노출되었을 가능성과 과거 및 현재 면역억제요법에 대한 자세한 의학적 평가가 포함되어야 한다. 모든 환자에서 투베르쿨린 피부 검사와 흉부 X ‑선과 같은 적절한 스크리닝 시험을 실시해야 한다(국가별 권장사항을 적용할 수 있음).
특히 결핵이 널리 유행되는 지역에서 이민오거나 혹은 그곳을 여행한 환자들 또는 활동성 결핵 환자와 긴밀한 접촉이 있었던 환자들에서 진단되지 않은 잠복성 결핵이 가능하다는 것을 고려해야만 한다.
활동성 결핵으로 진단된 경우에는 이 약의 투여를 시작해서는 안 된다.
잠복성 결핵으로 진단된 경우에는, 이 약의 투여 시작 전 항결핵 예방요법과 함께 적절한 치료를 시작해야 하며, 국가별 권장사항에 따라야 한다. 결핵 테스트 결과가 음성일지라도 결핵의 중대한 위험인자를 갖고 있거나 여러 위험인자가 있는 환자 및 과거 활성 또는 잠복성 결핵의 병력이 있으나 적절한 치료가 확인되지 않은 환자 또한 이 약의 투여 시작 전에 항결핵 예방요법을 고려해야한다. 이 환자에 대한 항결핵 치료 시작은 잠복성 결핵 감염에 대한 위험과 항결핵 치료의 위험 두 가지를 모두 고려한 후에 결정되어야만 한다. 만약 필요하다면, 결핵치료의 경험이 있는 의사와 상담하여야만 한다.
잠복성 결핵 감염 환자의 항결핵 치료는 이 약을 투여받는 환자들의 결핵 재활성화 위험을 감소시킨다. 항결핵 예방요법에도 불구하고, 이 약을 투여 받는 동안 활동성 결핵이 발생한 경우가 있었다. 또한 잠복성 결핵 결과가 음성이었던 환자에서 이 약을 투여받고 활성 결핵이 발생하고, 활성 결핵 치료가 성공적으로 실시된 몇몇 환자에서도 TNF 저해제로 투여받는 동안 활성 결핵이 다시 나타난 바가 있다.
이 약으로 치료받는 환자는 잠복성 결핵이 위음성일 수 있으므로, 활성 결핵의 증상 및 징후가 모니터링되어야 한다. 투베르쿨린 위음성(false negative) 결과의 위험은 중증 질환자나 면역기능저하환자에서 특히 고려해야한다.
만약 이 약으로 치료하는 동안이나 치료 후 결핵을 암시하는 증상/징후(예. 지속성 기침, 쇠약/체중감소, 미열, 무기력)가 나타나는 경우 의사에게 즉시 알리도록 환자를 교육해야 한다.
⓷ 기타 기회감염
침습적 진균 감염을 포함한 기회감염이 이 약을 투여 받고 있는 환자들에서 관찰되었다. 이 감염들은 TNF 저해제를 투여 받고 있는 환자들에게서 일관되게 나타나지는 않았으며, 이는 적절한 치료의 지연을 야기하였고, 때로는 치명적인 결과를 초래하였다.
TNF 저해제를 투여받고 있는 환자들은 히스토플라스마증, 콕시디오이데스진균증, 또는 분아균증(blastomycosis), 아스페르길루스증, 칸디다증 및 기타 기회감염들에 좀 더 감수성이 높다. 발열, 권태감, 체중 감소, 발한, 기침, 호흡곤란 및/또는 폐 침윤, 또는 기타 심각한 (쇼크를 동반하거나 동반하지 않은) 전신질환이 나타난 환자는 즉시 진단 평가를 위한 의학적 조치를 취해야 한다.
진균증이 풍토병인 지역에 머물거나 여행한 환자의 경우, 전신적인 진균 감염의 징후가 나타나면, 침습적인 진균 감염을 의심해야 한다. 환자들은 히스토플라스마증 및 기타 침습적 진균 감염의 위험이 있으므로, 임상 의사들은 병원균이 확인되기 전까지 실험적 항진균 치료를 고려해야 한다. 활성 감염 상태인 몇몇의 환자들은 히스토플라스마증의 항원 및 항체 검사에서 음성을 나타낼 수 있다. 이러한 환자들에게 실험적 항진균 치료제의 투여는, 침습적 진균 감염의 치료 및 진단에 전문가인 의사와 상담하여 결정되어야 하며, 심각한 진균 감염의 위험 및 항진균 치료의 위험을 모두 고려하여야 한다. 심각한 진균 감염이 발생한 환자 또한 감염이 조절되기 전까지 TNF 저해제의 사용 중단이 고려되어야 한다.2) 악성종양과 림프증식성 질환
TNF 저해제에 대한 임상시험의 대조 시험동안 TNF 저해제를 투여받은 환자에서 대조군 환자에 비해 더 많은 림프종 사례가 관찰되었다. 그러나 발생이 드물었고 위약군 환자의 추적조사기간이 TNF 저해제를 투여받은 환자에 비해 더 짧았다. 더욱이 오래 지속된, 매우 활동성인, 염증성 질환을 가진 류마티스 관절염 환자는 림프종의 기저 위험(background risk)이 증가되어 있고 이는 위험 추정을 어렵게 한다. 이 약을 이용한 장기간 공개 임상시험에서 악성종양의 전체 발현율은 나이, 성별, 인종이 맞춰진 전체 인구에서 예상되는 발현율과 유사했다. 현재의 지식으로 TNF 저해제를 투여받는 환자에서 림프종 또는 다른 악성종양의 발생 위험 가능성을 배제할 수 없다.
TNF 저해제를 투여받는 어린이와 청소년들에서 악성종양이 보고되었고, 일부는 치명적이었다. 이 보고된 경우 중 대략 절반의 경우는 호지킨 및 비호지킨 림프종을 포함한 림프종이었다. 나머지 다른 경우들에서는 여러 다양한 악성 종양들이 나타났으며, 일반적으로 면역억제와 연관되어 드물게 나타나는 악성종양도 포함되었다. 악성종양은 중간값으로 치료 30개월 후에 나타났으며, 대부분의 환자가 면역억제제를 병용 투여하고 있었다. 이는 시판 후 조사에서 보고되었으며, 등록과 자발적인 시판 후 보고서를 포함한 여러 경로에서 나타났다.
이 약을 투여받은 환자에서, Hepatosplenic T ‑cell lymphoma(HSTCL)가 시판후 조사에서 매우 드물게 보고되었고, 공격적 림프종(종종 치명적임)이 드물게 확인되었다. 대부분의 환자들은 염증성대장질환의 치료를 위하여 아자티오프린 또는 6 ‑메르캅토푸린의 병용 투여는 물론이고 이전에 인플리시맵으로 치료를 받았었다. 이 약과 아자티오프린 또는 6 ‑메르캅토푸린의 병용 투여 시의 잠재적인 위험성을 주의깊게 고려해야 한다. 이 약 복용환자에서 HSTCL의 발생 위험 가능성은 배제할 수 없다
악성종양의 병력이 있는 환자를 포함하거나 이 약 투여 중 악성종양이 발생한 환자에게 투여를 지속한 시험은 실시되지 않았다. 따라서 이러한 환자군에 투여를 고려시 추가적인 주의를 기울여야 한다. 모든 환자, 특히 과다한 면역억제제 치료의 병력이 있는 환자나, PUVA 치료의 병력이 있는 건선 환자들은 이 약의 투여 시작 전 및 투여 기간 동안 비흑색종 피부암 존재 여부에 대한 검사를 받아야 한다.
TNF 저해제를 류마티스 관절염과 다른 적응증에서 사용하여 급성 및 만성 백혈병이 나타난 경우가 보고되었다. 류마티스 관절염이 있는 환자들은 TNF 저해제를 투여받지 않더라도 다른 사람들에 비해 백혈병이 발병될 확률이 (2배까지) 높을 수 있다.
중등증에서 중증의 만성 폐쇄성 폐질환(COPD) 환자들을 대상으로 다른 TNF 저해제인 인플릭시맵을 사용하여 실시한 탐색적 임상시험에서, 현재 흡연중이거나 기존에 흡연하였던 사람의 경우 비교군의 환자보다 인플릭시맵 치료를 받은 환자군에서 악성 종양이 많이 보고되었다. 과도한 흡연으로 인해 악성종양의 위험성이 증가한 환자는 물론 만성 폐쇄성 폐질환(COPD) 환자에 대한 TNF ‑α 저해제 치료 고려 시 주의가 필요하다.
이 약이 형성이상증이나 대장암의 발생 위험에 영향을 미치는지 여부는 현재 알려지지 않았다. 형성이상증 또는 대장암종의 위험이 증가되었거나(예, 오래 지속되는 궤양성 대장염 또는 원발성 경화성 담관염), 형성이상증 또는 대장암종의 병력이 있는 모든 궤양성 대장염 환자들은 치료 시작 전 및 치료 중에 주기적으로 형성이상증에 대해 확인해야한다. 대장 내시경검사 및 생검 등 국가별 권장사항에 따라 평가한다.
3) B형 간염 재활성화
이 약을 포함한 TNF 저해제의 사용으로 B형 간염 만성 보균자에서 병증이 재활성화 될 수 있으며, 일부는 치명적이었다. 이 보고내용의 대부분은 다른 면역억제제를 함께 투여받는 환자에게서 나타났으며, 이는 또한 B형 간염 재활성화를 일으킬 수 있다. B형 간염의 감염 위험이 있는 환자는 이 약을 투여 받기 전 감염 여부에 대한 검사를 하여야 한다. B형 간염 보균자로 확인된 환자에게 TNF 저해제를 처방하는 경우, 의사는 주의를 기울여야 한다. B형 간염 보균자이고, TNF 저해제의 치료를 요하는 환자는 이 약을 투여 받는 동안과 투여가 끝난 후 수 개월 동안 감염의 증상/징후를 철저히 관찰해야 한다. B형 간염 보균자의 재활성화를 방지하기 위해 항바이러스제와 TNF 저해제를 함께 사용하는 것에 대한 적절한 치료법은 알려진 바 없다. B형 간염이 재활성화 된 경우 이 약의 투여를 중지하고 적절한 치료와 함께 항바이러스제 치료를 시작해야 한다.
4) 신경학적 반응
이 약을 포함한 다른 TNF 저해제는 드물게, 다발성 경화증 및 시신경염을 포함한 중추신경계 탈수초성질환(demyelinating disease)과 길랑 바레 증후군(Guillian Barre syndrome)을 포함한 말초 탈수초성질환의 임상 증상 및/또는 방사선 증거의 새로운 발현 혹은 악화와 관련이 있다. 중추신경계 및 말초신경계 탈수초성질환의 기왕력이 있거나 최근에 발현된 환자에게 이 약의 투여를 고려하는 경우 주의를 기울여야 한다. 이러한 이상이 진행되면 이 약의 중단을 고려하여야 한다.
중등도 포도막염과 중추 신경계 탈수초성장애 간의 연관성은 알려져있다. 비 ‑감염성 중등도 포도막염 환자에서 이 약 치료 전 중추신경계 탈수초성 장애 여부를 측정하는 신경학적 평가를 하여야 한다.
2. 다음 환자에는 투여하지 말 것
1) 이 약 또는 이 약의 성분에 과민증인 환자
2) 활동성 결핵 또는 패혈증, 기회감염과 같은 다른 중증 감염이 있는 환자(경고항 참조)
3) 중등도 내지 중증의 심부전(NYHA class III/IV) 환자
3. 이상반응
1) 이 약은 치료적 확증 대조 및 공개 임상시험을 통해 60개월 이상에 걸쳐 9,506명의 환자에 대하여 연구되었다. 이 임상시험들에는 단기 및 장기 질병상태를 보이는 류마티스 관절염 환자뿐만 아니라 건선성 관절염 및 축성 척추관절염(강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염) 환자 및 크론병 환자, 궤양성 대장염 환자 및 건선 환자, 화농성 한선염 환자, 포도막염 환자와 소아 특발성 관절염 (다관절 소아 특발성 관절염 및 골부착부위염 관련 관절염) 환자가 포함되었다. 표 1의 자료는 대조 임상시험 기간 동안 이 약을 투여받은 6,089명의 환자와 위약 또는 활성 대조약을 투여받은 3,801명의 환자를 포함한 주요 대조 임상시험 결과를 근거로 한 것이다. 주요 임상시험 중 이중 눈가림, 대조 임상시험 기간 동안 이상사례로 인해 투여를 중단한 환자의 비율은 이 약 투여군이 5.9%이고 대조군이 5.4%였다.
일반적으로 소아환자에서의 이상사례는 성인 환자에서 나타난 이상사례의 빈도 및 타입과 유사하였다.
이 약을 이용한 대조 임상시험에서 가장 빈번히 나타났던 이상사례들 중 하나를 근거로 약 13%의 환자들이 주사부위반응을 경험할 것으로 예상할 수 있다.
아래 표 1은 주요 임상시험에서 임상적 및 실험실 검사 모두 적어도 이 약과 인과관계의 가능성이 있는 약물이상반응을 발현부위와 빈도별(매우 자주 ‑1/10 이상 ; 자주 ‑1/100 이상 및 1/10 미만 ; 때때로 ‑1/1,000 이상 및 1/100 미만 ; 드물게 ‑1/1,000 미만)로 요약하였다. 각각의 발현빈도 그룹 안에, 약물이상반응은 심각성이 감소하는 순서로 배열되었다. 이 약의 여러 적응증에서 가장 자주 나타난 약물이상반응들이 포함되었다. 신체기관 열에서 (*)로 표시된 부분은 1. 경고, 4. 일반적 주의 항에서 자세한 내용을 확인할 수 있다.
2) 베체트 장염 : 일본의 베체트 장염 연구에서의 안전성 프로파일은 이미 연구된 이 약의 안전성 프로파일과 유사하였다.
3) 화농성 한선염: 이 약으로 매주 치료받은 화농성 한선염 환자의 안전성 프로파일은 이미 알려진 이 약의 안전성 프로파일과 유사하였다.
4) 포도막염: 이 약으로 치료받은 비 ‑감염성 포도막염 환자의 안정성 프로파일은 이미 알려진 이 약의 안전성 프로파일과 유사하였다.
<표 1. 임상시험 중 약물이상반응> <! ‑ ‑표 ‑ ‑><! ‑ ‑표 ‑ ‑><! ‑ ‑표 ‑ ‑>
|
신체기관 |
빈도 |
약물이상반응 |
|
감염 |
매우 자주 |
기도 감염(상기도 및 하기도 감염, 폐렴, 부비동염, 인두염, 비인두염, 폐렴 헤르페스 바이러스 포함) |
|
자주 |
전신 감염(패혈증, 칸디다증, 인플루엔자 포함), 장내 감염(위장염바이러스 포함), 피부 및 연조직 감염(손발톱주위염, 연조직염, 괴사성 근막염, 바이러스성 수막염, 게실염, 상처 감염, 농가진, 괴사근막염, 대상포진), 귀 감염, 구내감염(단순포진, 구강 헤르페스, 치아 감염 포함), 생식기 감염(진균성 외음부 및 질 감염 포함), 비뇨기계 감염(신우신염 포함), 진균 감염, 관절 감염 | |
|
때때로 |
기회감염과 결핵(콕시디오이데스진균증, 히스토플라스마증, 계형결핵균 복합체 감염), 신경 감염(바이러스성 수막염 포함), 눈 감염, 박테리아 감염 | |
|
양성, 악성 및 미확인 신생물(낭종 및 폴립 포함)(*) |
자주 |
양성종양, 악성흑색종을 제외한 피부암(기저세포암, 편평세포암을 포함) |
|
때때로 |
림프종, 실질기관종양(유방암, 폐종양, 갑상선 종양 포함), 악성흑색종 | |
|
혈액계 및 림프계 이상(*) |
매우 자주 |
백혈구감소증(호중구감소증, 무과립구증 포함), 빈혈 |
|
자주 |
저혈소판증, 백혈구증가증 | |
|
때때로 |
특발성 혈소판감소성 자반증 | |
|
드물게 |
범혈구감소증 | |
|
면역계 이상 |
때때로 |
과민반응, 알레르기(계절성 알레르기 포함) |
|
대사 및 영양계 이상 |
매우 자주 |
지질증가 |
|
자주 |
저칼륨혈증, 고뇨산혈증, 혈중나트륨농도이상, 저칼슘혈증, 고혈당증, 저인산혈증, 탈수증 | |
|
정신계 이상 |
때때로 |
감정이상(우울증 포함), 불안, 불면증 |
|
신경계 이상 |
매우 자주 |
두통 |
|
자주 |
피부지각이상(감각저하증 포함), 편두통, 신경뿌리압박 | |
|
때때로 |
떨림, 신경병증 | |
|
드물게 |
다발성경화증 | |
|
눈의 이상 |
자주 |
시각장애, 결막염, 눈꺼풀염, 눈부종 |
|
때때로 |
복시 | |
|
귀 및 미로 이상 |
자주 |
어지럼증 |
|
때때로 |
난청, 이명 | |
|
심장 이상 |
자주 |
빈맥 |
|
때때로 |
부정맥, 울혈성 심부전 | |
|
드물게 |
심정지 | |
|
혈관 이상 |
자주 |
고혈압, 홍조, 혈종 |
|
때때로 |
혈관동맥 폐색증, 혈전정맥염, 대동맥류 | |
|
호흡, 흉부 및 종격 이상 |
자주 |
기침, 천식, 호흡곤란 |
|
때때로 |
만성 폐쇄성 폐질환, 간질성 폐질환, 폐렴 | |
|
위장관계 이상 |
매우 자주 |
복통, 구역 및 구토 |
|
자주 |
위장관출혈, 소화불량, 위식도역류 질환, 건조증후군 | |
|
때때로 |
췌장염, 연하곤란, 얼굴부종 | |
|
간 및 담즙계 |
매우 자주 |
간 효소 상승 |
|
때때로 |
담낭염, 담석증, 빌리루빈 상승, 지방간 | |
|
피부 및 피하조직 이상 |
매우 자주 |
발진(박리성 발진 포함) |
|
자주 |
가려움증, 두드러기, 타박상(자색반증 포함), 피부염(습진 포함), 조갑박리증, 다한증 | |
|
때때로 |
야간발한, 흉터 | |
|
근골격계 및 결합조직 이상 |
매우 자주 |
근골격통 |
|
자주 |
근경련(혈중크레아틴인산활성효소 증가 포함) | |
|
때때로 |
횡문근융해, 전신홍반루푸스 | |
|
신장 및 비뇨 이상 |
자주 |
혈뇨, 신기능 손상 |
|
때때로 |
야뇨증 | |
|
생식기계 및 유방 이상 |
때때로 |
발기부전 |
|
전신이상 및 투여 부위 이상 |
매우 자주 |
주사부위반응(주사부위 홍반 포함) |
|
자주 |
흉통, 부종 | |
|
때때로 |
염증 | |
|
임상 실험실 검사 |
자주 |
응고 및 출혈이상(활성화부분트롬보플라스틴시간 지연 포함), 자가항체시험 양성(이중나선DNA항체 포함), 혈중 락트산탈수효소 증가 |
|
상해 및 중독 |
자주 |
치유 부전 |
이 약으로 치료를 받은 류마티스 관절염 환자에게 5% 미만의 드문 발생률로 나타난 기타 중대한 이상사례는 다음과 같다.
전신 : 발열, 감염, 극심한 통증, 골반 통증, 패혈증, 수술, 가슴통증, 결핵의 재발
심혈관계 : 부정맥, 심방 세동, 심혈관 장애, 흉통, 울혈성 심부전, 관상동맥 이상, 심정지, 고혈압성 뇌병증, 심근경색증, 심계항진, 심낭삼출, 심낭염, 실신, 빈맥, 혈관 장애
콜라겐 질환 : 홍반성 루푸스 증후군
소화기계 : 담낭염, 담석증, 식도염, 위염, 위장장애, 위장출혈, 간 괴사, 구토
내분비계 : 부갑상선 장애
혈액계 : 무과립구증, 과립구감소증, 백혈구감소증, 림프종 유사반응, 범혈구감소증, 적혈구증가증
대사 및 영양장애 : 탈수증, 치유 이상, 케톤증, 파라프로테인혈증, 말초부종
근골격계 : 관절염, 뼈장애, 뼈골절(자연적인 경우가 아닌), 골괴사, 관절장애, 근육경련, 중증근무력증, 화농성 관절염, 윤활막염, 힘줄 장애
종양 : 샘종, 유방 ◦ 위장관 ◦ 피부 ◦ 비뇨생식과 같은 암종 및 기타(림프종, 흑색종)
신경계 : 착란, 다발성 경화증, 감각이상, 경막하혈종, 떨림
호흡기계 : 천식, 기관지연축, 호흡곤란, 폐 장애, 폐기능 감소, 흉막 삼출, 폐렴
피부 및 부속물 : 연조직염, 얕은 연조직염, 대상포진
특수감각 : 백내장
혈전증 : 다리혈전
비뇨생식기계 : 방광염, 신장 결석, 월경 장애, 신우신염
5) 주사부위반응 : 성인 및 소아에 대한 주요 대조 임상시험에서 이 약을 투여한 환자의 12.9%가 이 약을 투여했을 때 가장 흔히 나타난 이상사례인 주사부위반응(홍반 및/또는 가려움증, 출혈, 통증 또는 부종)을 나타낸 반면, 위약 또는 활성 대조군을 투여한 환자에서는 7.2%에서 이러한 반응이 나타났다. 주사부위반응은 일반적으로 투여 중단이 필요하지는 않았다.
6) 감염 : 성인 및 소아에 대한 주요 대조 임상시험에서, 감염률은 이 약 투여군이 1.51/환자 ‑년(patient year)이고 위약 및 활성 대조약 투여군은 1.46/환자 ‑년이었다. 감염은 주로 비인두염, 상기도 감염 및 부비동염으로 구성되었다. 대부분의 환자들은 감염이 소실된 후 이 약을 계속 투여하였다. 심각한 감염 발생률은 이 약 투여군이 0.04/환자 ‑년이고 위약 및 활성 대조약 투여군은 0.03/환자 ‑년이었다. 이 약을 이용한 성인 및 소아에 대한 대조 및 공개 임상시험에서 심각한 감염(드물게 발생한 치명적 감염을 포함)이 보고되었으며, 이는 결핵(속립성 및 폐 이외 부위 포함) 및 침습적 기회 감염(예. 파종성 히스토플라스마증, 폐포자충 폐렴, 아스페르길루스증 및 리스테리아증)을 포함한다. 결핵 사례의 대부분은 치료 시작 후 최초 8개월 이내에 발생하였으며, 이는 잠복 질환의 재발을 반영하는 듯하다.
7) 악성종양과 림프증식성 질환 : 소아 특발성 관절염(다관절형 소아 특발성 관절염 및 골부착부위염 관련 관절염) 환자를 대상으로 655.6 환자 ‑년 노출된 249명에서의 임상시험 동안 악성종양이 관찰되지 않았다. 또한, 소아 크론병 환자를 대상으로 498.1환자 ‑년 노출된 192명에서의 임상시험 동안 악성종양이 관찰되지 않았다.
판상 건선 소아 환자를 대상으로 80.0 환자 ‑년 노출된 77명의 소아 환자에서의 임상시험 동안 악성 종양이 관찰되지 않았다. 중등도에서 중증의 류마티스 관절염, 건선성 관절염, 축성 척추관절염(강직성 척추염 및 방사선학적으로 강직성 척추염이 확인되지 않는 중증 축성 척추관절염), 크론병, 궤양성대장염, 화농성 한선염, 건선, 포도막염 환자를 대상으로 12주 이상 실시한 성인에 대한 주요 대조 임상시험에서 림프종, 비흑색종 피부암 이외에도 악성종양이 투여군 5,291명 중에서 6.8/1,000 환자 ‑년, 대조군 3,444명 중에서 6.3/1,000 환자 ‑년의 비율로 생성되었다(치료기간의 중간값은 이 약의 투여군에서는 4.0개월, 대조군에서는 3.8개월이었다). 비흑색종 피부암은 투여군에서 8.8/1,000 환자 ‑년 및 대조군에서 3.2/1,000 환자 ‑년의 비율로 생성되었다. 피부암중에서 편평세포암종은 투여군에서 2.7/1,000 환자 ‑년, 대조군에서 0.6/1,000 환자 ‑년의 비율로 생성되었다. 림프종의 비율은 투여군에서 0.7/1,000 환자 ‑년, 대조군에서 0.6/1,000 환자 ‑년이었다.
대조 임상시험과 진행 중 또는 완료된 공개 연장 시험에서, 림프종 및 비흑색종 피부암 이외에 관찰된 악성종양의 발생률은 약 8.5/1,000 환자 ‑년이다. 비흑색종 피부암의 관찰된 비율은 약 9.6/1,000 환자 ‑년이며, 림프종이 관찰된 비율은 약 1.3/1,000 환자 ‑년이다. 이 임상시험기간의 중앙값은 약 3.3년이며, 최소 1년 동안 이 약을 투여받은 6,427명의 환자들 또는 26,439.6 환자 ‑년 투여를 초과하는 치료 시작 1년 이내에 악성종양으로 발전한 환자들을 포함했다.
2003년 1월부터 2010년 12월까지의 시판 후 경험 중 주로 류마티스 관절염 환자에서 보고된 악성종양의 발생 비율은 약 2.7/1,000 환자 ‑년 이었다. 비흑색종 피부암과 림프종의 발생 비율은 각각 약 0.2 및 0.3/1,000 환자 ‑년이었다.
8) 자가항체 : 류마티스 관절염에 대한 5건의 임상시험 중 여러 시점에서 혈청 내 자가항체에 대한 검사를 실시했다. 이러한 적절히 디자인된 대조 임상시험에서 기저 항 핵항체(ANA, anti ‑nuclear antibody)가 음성이었던 환자 중 이 약 투여군의 11.9%와 위약 및 활성 대조군의 8.1%에서 24주째 양성을 나타냈다. 모든 류마티스 관절염과 건선성 관절염에 대해 이 약을 투여받은 3,441명 중 2명에서 새롭게 발생된 루푸스양 증후군임을 시사하는 임상 증상이 나타났으며 이 환자는 투여 중단 후 개선되었다. 루푸스성 신장염이나 중추신경계 증상이 나타난 환자는 없었다.
9) 건선 : 발현 및 악화
이 약을 포함하여 TNF 저해제를 사용 시, 농포성 건선 및 손발바닥 건선을 포함하여 건선이 첫 발현되는 경우와 이미 존재하던 건선이 악화되는 경우들이 보고되었다. 이 환자들의 대다수는 면역억제제(예. MTX, 코르티코스테로이드)를 병용하고 있었으며, 일부는 입원치료가 필요했다. 대부분의 환자는 TNF 저해제의 복용을 중단함으로써 건선이 호전되었다. 일부 환자들은 이후, 다른 TNF 저해제를 재 투여했을 때 건선이 재발하였다. 국소 치료에도 불구하고 증상이 호전되지 않거나, 악화되는 심각한 경우에는 이 약의 투여 중지를 고려해야만 한다.
10) 간 효소 상승
◦ 류마티스 관절염 임상시험 : 류마티스 관절염에 대한 대조 임상시험에서(4건의 임상시험), ALT의 상승은 이 약 또는 위약을 투여한 환자에서 유사했다. 초기 류마티스 관절염 환자에서(3년 미만의 유병기간)(1건의 임상시험), ALT의 상승은 병용 투여군(이 약/메토트렉세이트)에서 메토트렉세이트 단독군 또는 이 약 단독군에 비해 더 자주 발생했다. 다관절형 소아 특발성 관절염 환자를 대상으로 한 임상시험에서 일부 트랜스아미나제의 상승이 나타났으나 위약과 이 약 투여군에서 같거나 유사했으며, 대부분은 메토트렉세이트와 병용 투여 시 나타났다.
◦ 건선성 관절염 임상시험 : ALT의 상승은 류마티스 관절염 임상시험의 환자에 비해 건선성 관절염 환자에 대한 임상시험(2건의 임상시험)에서 더 자주 발생하였다.
◦ 모든 류마티스 관절염, 다관절형 소아 특발성 관절염 및 건선성 관절염 임상시험에서, 상승된 ALT를 나타내는 환자들은 증상이 없었고, 대부분 상승이 일시적이며 투여 지속시 소실되었다.
◦ 크론병 및 궤양성 대장염 임상시험 : 대조 임상시험에서 이 약 투여군과 대조군간의 ALT상승은 유사하였다.
체중에 따라 조정된 유도 요법 후 체중에 따라 조정된 유지 요법(최대 52주)의 유효성과 안전성을 평가한 소아 크론병의 3상 임상시험에서, 시험대상자의 2.6%(5/192)에서 ≧ 3 X ULN의 ALT 상승이 발생하였고, 이 중 4명은 베이스라인에서 면역억제제를 병용 투여하였다.
◦ 건선 임상시험 : 건선에 대한 대조 임상시험에서, 투여군과 대조군간의 ALT 상승은 유사하였다.
◦ 화농성 한선염 임상시험 : 화농선 한선염에 대한 대조 임상시험에서, 투여군의 0.3%와 대조군의 0.6%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다.
◦ 소아 특발성 관절염 임상시험 : 4 ‑ 17세 다관절형 소아 특발성 관절염 환자와 6 ‑ 17세 골부착부위염 관절염 환자의 3상 대조 임상 시험에서, 투여군의 6.1%와 대조군의 1.3%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다. 대부분의 ALT 상승은 메토트렉세이트 병용 사용에 따라 발생하였다. 2세에서 4세 미만의 다관절형 소아 특발성 관절염 환자에 대한 3상 임상시험에서, ≧ 3 X ULN의 ALT 상승은 발생하지 않았다.
◦ 판상 건선 임상시험 : 판상 건선 소아 환자에 대한 3상 임상시험에서 ≧ 3 X ULN의 ALT 상승은 발생하지 않았다.
◦ 포도막염 임상시험: 포도막염에 대한 대조 임상시험에서, 투여군의 2.4%와 대조군의 2.4%에서 ≧ 3 X ULN의 ALT 상승이 발생하였다.
모든 적응증의 임상시험에서 상승된 ALT를 나타내는 환자들은 증상이 없었고, 대부분 상승이 일시적이며 투여 지속 시 소실되었다. 그러나 간부전을 포함한 중증의 간손상이 이 약을 포함한 TNF 저해제를 투여받은 환자의 시판 후 보고에서 아주 드물게 나타났다. 이 약 투여와의 관계는 명백하지 않다.
11) 아자티오프린 또는 6 ‑메르캅토푸린과의 병용 투여 : 성인 크론병 임상시험에서, 아자티오프린/6 ‑메르캅토푸린과 병용 투여 시 이 약의 단독 투여에 비해 악성 종양 및 심각한 감염과 관련된 이상사례의 발생률이 더 높게 나타났다.
12) 시판 후 조사 또는 4상 시험에서 보고된 추가 이상사례 : 4상 시험과 시판 후 조사에서 보고된 추가 이상사례는 다음 표 2와 같다.
<표 2. 시판 후 조사 및 제4상 시험에서의 이상사례>
|
신체기관 |
이상사례 |
|
감염 및 기생충 감염 |
게실염 |
|
간담도계 이상* |
B형 간염의 재활성화, 간부전, 간염, 자가 면역성 간염 |
|
신경계 이상* |
탈수초성 장애(예. 시신경염), 길랑-바레 증후군, 뇌혈관사고 |
|
호흡, 흉부 및 종격계 이상 |
폐색전증, 흉막삼출, 폐섬유증 |
|
피부 및 피하 조직 이상 |
피부 혈관염, 스티븐스-존슨 증후군, 혈관부종, 건선의 발생 또는 악화(손발바닥 농포성 건선 포함), 다형홍반, 탈모, 태선모양 피부반응** |
|
면역계 이상* |
아나필락시스, 사르코이드증 |
|
위장관계 이상* |
장 천공 |
|
양성, 악성 및 미확인 신생물* |
Hepatosplenic T-cell lymphoma, 백혈병, 메르켈세포암(피부 신경내분비 선암) |
|
근골격계 및 결합조직 이상 |
루푸스 유사 증후군 |
|
심장 이상 |
심근경색증 |
|
전신 및 투여부위 이상 |
발열 |
|
* 1. 경고, 4. 일반적 주의 항에서 자세한 내용을 확인할 수 있다. ** 이 약을 포함한 TNF 저해제를 투여 받고 있는 환자들에게서 발생하였다. | |
⓵ 국내에서 6년 동안 류마티스 관절염, 건선성 관절염, 강직성 척추염, 크론병, 건선환자를 대상으로 실시한 시판 후 조사
국내에서 1,698명(류마티스 관절염 652명, 건선성 관절염 47명, 강직성 척추염 925명, 크론병 73명, 건선 1명)을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 10.07%(171명/1,698명, 238건)이었고, 이 중 이 약과 인과관계를 배제할 수 없는 약물이상반응 발현율은 6.83%(116명/1,698명, 141건)이며, 주사부위 반응 19건(1.11%), 가려움(증) 9건(0.53%), 발진 8건(0.47%), 대상포진, 상기도 감염, 두드러기 각 4건(0.24%), 신우신염, 홍반, 복통, 어지러움, 두통 각 3건(0.18%), 연조직염, 폐결핵, 결핵성 복막염, 폐렴, 결핵, 피부반응, 알레르기성 피부염, 림프종, 근육통, 기침, 림프절병증 각 2건(0.12%), 모낭염, 신장결핵, 비염, 편도염, 말라리아, 기관지 폐렴, B형 간염, 코 인두염, 감염성 윤활낭염, 진균성 감염, 크립토코쿠스 폐렴, 농포성 발진, 가려움 발진, 피부염, 탈모(증), 피부병변, 무력(증), 구토, 설사, 복막염, 구역, 고창, 갑상선암, 기관지 신생물, 급성 골수성 백혈병, 골관절염, 관절종창, 폐출혈, 콧물, 간손상, 간독성, 간기능 이상, 찔림 알레르기, 현기증, 월경 장애, 포도막염 각 1건(0.06%)이 보고되었다.
중대한 이상사례 발현율은 2.77%(47명/1,698명, 63건)이며, 신우신염, 복통이 각 4건이었으며 폐렴, 폐 결핵, 대상포진, 결핵성 복막염, 결핵, 설사, 구토, 가려움(증)이 각각 2건이며 그 외 신장결핵, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 항문 농양, 복막 농양, 결장염, 장관 천공, 복막염, 장관 경색, 장피부누공, 구역, 홍반, 발진, 대뇌출혈, 마비, 치매, 골관절염, 강직성 척추염, 등 통증, 갑상선암, 급성 골수성 백혈병, 직장암, 무력(증), 발열, 통증, 추골 탈구, 골절, 폐 출혈, 호흡곤란, 무릎 수술, 소장 절제(술), 혈중 크레아티닌 증가, 혈색소 감소, 간독성, 찔림 알레르기, 심근경색증, 빈혈, 탈수는 각 1건이 보고되었다.
이 중 이 약과 인과관계를 배제할 수 없는 중대한 약물이상반응 발현율은 1.47%(25명/1,698명, 30건)이며, 신우신염 3건, 복통, 폐결핵, 대상포진, 결핵성 복막염, 결핵, 가려움(증) 각 2건, 폐렴, 신장결핵, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 복막염, 홍반, 발진, 골관절염, 갑상선암, 급성 골수성 백혈병, 폐출혈, 혈중 크레아티닌 증가, 간독성, 찔림 알레르기 각 1건이 보고되었다.
예상하지 못한 이상사례 발현율은 3.24%(55명/1,698명, 61건)이며, 설사, 콧물 각 5건, 관절 종창, 포도막염 각 3건, 등 통증, 사지 통증, 감각 이상, 무력(증), 림프절병증 각 2건, 결장염, 복막염, 장관 경색, 위장관 궤양, 장피부누공, 명치불쾌감, 치루, 고창, 모낭염, 비염, 편도염, 말라리아, 기관지 폐렴, 감염성 윤활낭염, 크립토코쿠스 폐렴, 농포성 발진, 항문 농양, 복막 농양, 경부 통증, 강직성 척추염, 대뇌출혈, 마비, 치매, 신경근병증, 기억장애, 폐 출혈, 눈마름, 압통, 종양 절제(술), 간독성, 추골 탈구, 현기증, 피부병변은 각각 1건이 보고되었다.
이 중 이 약과 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 1.24%(21명/1,698명, 21건)이며, 림프절병증은 2건, 설사, 복막염, 고창, 모낭염, 비염, 편도염, 말라리아, 기관지 폐렴, 감염성 윤활낭염, 크립토코쿠스 폐렴, 농포성 발진, 관절 종창, 콧물, 폐 출혈, 포도막염, 무력(증), 간독성, 현기증, 피부병변은 각각 1건이 보고되었다. 중대하고 예상하지 못한 이상사례 발현율은 1.12%(19명/1,698명, 20건)이며, 설사는 2건, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 항문 농양, 복막 농양, 결장염, 복막염, 장관 경색, 장피부누공, 대뇌출혈, 마비, 치매, 강직성 척추염, 등 통증, 무력(증), 추골 탈구, 폐 출혈, 간독성은 각 1건이 보고되었고, 중대하고 예상하지 못한 약물이상반응 발현율은 0.35%(6명/1,698명, 6건)이며, 복막염, 말라리아, 감염성 윤활낭염, 크립토코쿠스 폐렴, 폐 출혈, 간독성 각 1건이 보고되었다.
⓶ 국내에서 4년 동안 소아 특발성 관절염(다관절형 소아 특발성 관절염, 골부착부위염 관련 관절염) 환자 28명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 21.43%(6명/28명, 8건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 17.86% (5명/28명, 7건) 이며, 독감 10.71% (3명/28명, 3건) 등통증, 관절부기, 두드러기, 발열 각 3.57% (1명/28명, 1건)이 보고되었다. 중대한 이상사례 발현율은 3.57%(1명/28명, 1건)이며, 홍반이 3.57% (1명/28명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응은 확인되지 않았다.
예상하지 못한 이상사례 발현율은 3.57% (1명/28명, 1건)이었고, 이 1건은 관절부기였으며 이는 본 제와 인과관계를 배제할 수 없다. 중대하고 예상하지 못한 이상사례 및 중대하고 예상하지 못한 약물이상반응은 확인되지 않았다.
⓷ 국내에서 4년 동안 소아 크론병 환자 143명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 18.18%(26명/143명, 47건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 14.69%(21명/143명, 26건)이며, 백혈구감소증 2.80%(4명/143명, 4건), 발진 2.10%(3명/143명, 3건), 설사, ALT증가, AST증가 각 1.40%(2명/143명, 2건), 대변내혈액, 마비성장폐쇄, 장천공, 가려움증, 국소피부반응, 두드러기, 상세불명의간기능검사이상, 충수돌기염, 권태, 주사부위홍반, 칸디다증, 신우신염, 모낭염이 각 0.70%(1명/143명, 1건) 보고되었다.
중대한 이상사례 발현율은 5.59%(8명/143명, 13건)이며, 복통 1.40%(2명/143명, 2건), 창자괴사, 대변내혈액, 마비성장폐쇄, 장폐쇄, 장천공, 소장폐쇄, 충수돌기염, 복막염, 열, 칸디다증, 신우신염이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응 발현율은 3.50%(5명/143명, 5건)이며, 마비성장폐쇄, 장천공, 충수돌기염, 칸디다증, 신우신염이 각 0.70%(1명/143명, 1건) 보고되었다.
예상하지 못한 이상사례 발현율은 6.29%(9명/143명, 17건)이며, AST증가 2.10%(3명/143명, 3건), 창자협착증 1.40%(2명/143명, 2건), 대장혈종, 위장염, 창자괴사, 마비성장폐쇄, 장폐쇄, 소장폐쇄, 충수돌기염, 헬리코박터파이로리위염, C반응단백질증가, 적혈구침강속도증가, 난치성통증, 명시안된수술후합병증이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 2.80%(4명/143명, 4건)이며, AST증가 1.40%(2명/143명, 2건), 마비성장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다.
중대하고 예상하지 못한 이상사례 발현율은 3.50%(5명/143명, 5건)이며, 창자괴사, 마비성장폐쇄, 장폐쇄, 소장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 중대하고 예상하지 못한 약물이상반응 발현율은 1.40%(2명/143명, 2건), 마비성장폐쇄, 충수돌기염이 각 0.70%(1명/143명, 1건) 보고되었다.
⓸ 국내에서 4년 동안 성인 화농성 한선염 환자 및 소아 판상 건선 환자 19명을 대상으로 실시한 시판 후 사용성적조사 결과, 이상사례 발현율은 42.11%(8명/19명, 12건)이었고, 이 중 본 제와 인과관계를 배제할 수 없는 약물이상반응 발현율은 31.58%(6명/19명, 10건)이며, 한선염 15.79%(3명/19명, 4건), 여드름 5.26%(1명/19명, 2건), 소양증, 종기, 편도염, 발열이 각각 5.26%(1명/19명, 1건)으로 보고되었다. 중대한 이상사례 및 본 제와 인과관계를 배제할 수 없는 중대한 약물이상반응은 확인되지 않았다. 예상하지 못한 이상사례 발현율은 21.05%(4명/19명, 6건)이었고, 한선염 15.79%(3명/19명, 4건), 여드름 5.26%(1명/19명, 2건)으로 보고되었다. 이 중 본 제와 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응 발현율은 21.05%(4명/19명, 6건)이었다. 중대하고 예상하지 못한 이상사례 및 중대하고 예상하지 못한 약물이상반응은 확인되지 않았다.
⓹ 국내에서 포도막염에 대해 재심사를 위하여 4년 동안 155명을 대상으로 실시한 시판 후 조사 결과, 이상사례의 발현율은 인과관계와 상관없이 8.39%(13명/155명, 총 25건)로 보고되었다. 이 중 이 약과 인과관계를 배제할 수 없는 약물이상반응 발현율은 3.23%(5명/155명, 6건)이며, 안구 불편감, 주사부위 과민증, 주사 부위 통증, 지각 이상, 근육통, 습진 각 0.65%(1명/155명, 1건) 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응은 없었고, 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 1.29%(2명/155명, 2건) 안구 불편감, 주사부위 과민증 각 0.65%(1명/155명, 1건) 보고되었다.
➅ 이 약에 대한 국내 재심사 이상사례 및 자발적 부작용 보고자료를 국내 시판 허가된 모든 의약품을 대상으로 보고된 이상사례 보고자료(1989 ‑2021.5.31)와 재심사 종료시점에서 통합평가한 결과, 다른 모든 의약품에서 보고된 이상사례에 비해 이 약에서 통계적으로 유의하게 많이 보고된 이상사례 중 새로 확인된 것들은 다음과 같다. 다만, 이 결과가 해당성분과 다음의 이상사례 간에 인과관계를 입증된 것을 의미하는 것은 아니다.
◦ 근육 ‑골격계 이상 : 관절통, 관절병증
◦ 대사 및 영양계 이상 : 체중증가, 고지혈증
◦ 전신 및 투여부위 이상 : C반응단백질증가, 주사부위 덩어리, 주사부위 멍듦, 상태악화
◦ 호흡기계 이상 : 가래증가
◦ 정신계 이상 : 식욕증가
◦ 중추 및 말초신경계 이상 : 보행이상
◦ 생식기계 이상(여성) : 월경과다
◦ 소화기계 이상 : 흑색변
◦ 감염 : 농양, 종기증
◦ 피부 및 피하조직계 이상 : 각화과다증
14) 국내 관찰연구 결과
국내에서 베체트장염에 대해 6년 동안 50명을 대상으로 관찰 연구한 결과, 이상사례의 발현율은 인과관계와 상관없이 72%(36/50, 총 122건)로 보고되었다. 이 중 인과관계를 배제할 수 없는 중대한 약물이상반응 및 인과관계를 배제할 수 없는 예상하지 못한 약물이상반응은 발현 빈도에 따라 아래 표에 나열하였다.
|
|
중대한 약물이상반응 8%(4명/50명, 11건) |
예상하지 못한 약물이상반응 14%(7명/50명, 9건) | |
|
때때로(0.1~5%미만) |
각종 위장관 장애 |
복통, 상복부 통증, 설사, 오심 |
입 궤양 형성, 베체트 증후군, 상복부 통증, 변비 |
|
감염 및 기생충 감염 |
급성 신우신염, 파종 결핵, 인두염 |
| |
|
각종 정신 장애 |
|
수면 장애 | |
|
전신 장애 및 투여 부위 병태 |
발열 |
주사 부위 경화 | |
|
대사 및 영양 장애 |
섭식 저하 |
섭식 저하 | |
|
호흡기, 흉곽 및 종격 장애 |
기침 |
| |
|
피부 및 피하 조직 장애 |
|
접촉 피부염 | |
1) 알레르기 반응 : 임상시험 동안 이 약의 피하주사로 인한 심각한 알레르기 반응은 드물게 보고되었다. 이 약 투여 후 아나필락시스를 포함한 중대한 알레르기 반응이 보고된 바 있다. 만일 아나필락시스 반응이나 다른 심각한 알레르기 반응이 나타나면 이 약의 투여를 즉시 중단하고 적절한 치료를 시작해야 한다.
2) 면역억제 : 이 약을 투여받은 64명의 류마티스 관절염 환자에 대한 시험에서 지연성 과민반응의 억제, 면역글로불린 억제 또는 effector T ‑와 B세포, NK ‑세포, 단핵세포/대식세포 및 호중구의 수적 변화의 증거는 없었다.
3) 혈액학적 반응 : TNF 저해제에서 드물게 재생불량성빈혈을 포함한 범혈구감소증이 보고되었다. 의학적으로 중요한 혈구감소증(예. 저혈소판혈증, 백혈구감소증)을 포함한 혈액계의 이상사례가 이 약에서 보고되었다. 이러한 보고의 이 약에 대한 원인 상관관계는 불분명하다. 이 약을 투여하는 동안 혈액질환을 암시하는 징후 또는 증상(예, 지속적인 발열, 타박상, 출혈, 창백)이 발현되는 경우 즉각적인 의학적 처치를 받도록 모든 환자를 교육해야 한다. 심각한 혈액학적 이상이 확정된 환자는 이 약의 투여 중단을 고려해야 한다.
4) 예방접종 : 이 약 또는 위약을 투여한 226명의 성인 류마티스 관절염 환자에 대한 시험에서 표준 23가 폐구균 백신과 3가 인플루엔자 백신에 대한 항체반응과 유사한 반응이 관찰되었다. 이 약을 투여받은 환자에게 생백신을 투여한 경우 이에 의한 이차적인 감염 전파에 대한 이용 가능한 자료는 없다. 이 약을 투여 받고 있는 환자에게 생백신을 제외한 다른 백신을 병용 투여할 수 있다.
5) 울혈성 심부전 : 다른 TNF 저해제를 사용한 임상시험에서 울혈성 심부전의 악화와 울혈성 심부전으로 인한 사망률 증가가 관찰되었다. 이 약을 투여받은 환자에서도 울혈성 심부전이 악화된 사례가 보고되었다. 경증 심부전(NYHA classI/II) 환자에게 이 약 투여시 주의해야 하며 중등도 또는 중증 심부전 환자에게 투여해서는 안 된다. 울혈성 심부전이 새롭게 발생하거나 증상이 악화된 환자의 경우 이 약의 투여를 중단해야 한다.
6) 자가면역 과정 : 이 약의 투여로 자가면역 항체가 형성될 수 있다. 이 약의 장기투여가 자가면역질환 발생에 미치는 영향은 알려지지 않았다. 만일 이 약으로 치료 후 환자에게서 루푸스양 징후를 암시하는 증상을 나타내고 dsDNA에 대한 항체가 양성이라면 이 약의 투여를 중지해야 한다.
7) 생물학적 DMARDs 또는 TNF 저해제와의 병용투여 : 에타너셉트(다른 TNF 저해제)와 아나킨라를 병용 투여한 임상 시험에서 심각한 감염이 관찰되었으며, 에타너셉트 단독 요법에 비해 추가적인 유익성이 없었다. 에타너셉트와 아나킨라의 병용 시 관찰된 이상사례의 특성 때문에, 아나킨라와 다른 TNF 저해제와의 병용으로 인해 유사한 독성이 있을 수 있다. 이 약과 아나킨라의 병용 투여가 권장되지 않는다. 감염 및 기타 잠재적 약물학적 상호작용의 위험 증가 가능성으로 이 약과 다른 생물학적 DMARDs (예. 아나킨라 및 아바타셉트) 또는 다른 TNF 저해제와의 병용투여는 권장되지 않는다.
8) 수술 : 이 약을 투여받은 환자에서 수술 절차의 안전성 경험은 제한적이다. 만일 수술 절차가 계획된다면 이 약의 긴 반감기를 고려해야 한다. 이 약 투여동안 수술이 필요한 환자는 감염에 대해 면밀히 모니터링하고 적절한 조치를 취해야 한다. 이 약 투여 동안 관절성형술을 받는 환자에 대한 안전성 경험은 제한적이다.
9) 소장 폐색증 : 크론병의 치료에 반응하지 않은 환자는 외과적 치료를 요하는 고정된 섬유성 협착을 보일 수 있다. 이 약이 협착을 일으키거나 악화시키지 않는다는 것을 나타내는 자료가 있다.
10) 간 ◦신장애 환자 : 이러한 환자군에 대해서 연구되지 않았으며 권장 용량이 없다.
11) 이 약이 운전이나 기계 조작 능력에 미치는 영향에 대해서는 연구된 바 없다.
5. 상호작용
1) 이 약은 류마티스 관절염 환자에서 단독요법 및 메토트렉세이트와의 병용 요법 모두에 대해 연구되었다. 시험결과 이 약 또는 메토트렉세이트의 용량조절 필요성은 나타나지 않았다. 공식적인 약동학 시험에서 이 약과 메토트렉세이트 이외의 약물과의 상호작용을 평가하지 않았다. 임상시험에서 이 약과 일반적으로 사용되는 DMARDs(sulfasalazine, hydrochloroquine, leflunomide 및 parenteral gold), 글루코코르티코이드, 살리실산염, 비스테로이드성항염증제 또는 진통제를 병용 투여 시 상호작용이 나타나지 않았다.
이 약 단독투여에 비해 메토트렉세이트와 병용 투여 시 항체 생성률이 더 낮았다. 메토트렉세이트를 투여하지 않고 이 약 단독투여시 항체 생성률과 이 약의 제거율이 증가했으며, 이 약의 효과는 감소했다.
2) 생백신과 이 약의 병용 투여는 권장되지 않는다.
3) 아나킨라와 이 약의 병용 투여는 권장되지 않는다.
4) 약물/실험실 검사 상호작용 : 이 약과 실험실 검사 사이에 알려진 간섭은 없다.
6. 임부 및 수유부에 대한 투여
1) 원숭이에 대한 발생독성시험에서 모체 독성, 배아 독성 또는 최기형성이 나타나지 않았다. 출생후 독성과 수태능에 미치는 영향에 대한 비임상시험 자료는 없다. TNF 알파 저해 작용으로 인해 임신기간 중 이 약 투여는 신생아의 정상 면역반응에 영향을 줄 수 있다. 임신기간 동안 이 약은 태아에 대한 잠재적 유익성이 잠재적 위험성을 상회하는 경우에만 임부에게 투여해야한다. 임신할 가능성이 있는 여성은 임신을 예방하기 위한 적절한 피임요법 사용과 이 약의 최종 투여 후 최소 5개월간 피임을 지속할 것을 고려하여야 한다.
임신 중 노출에 대한 전향적 코호트 등록 연구에서, 임신 1기 동안 아달리무맙을 최소 1 회 투여한 류마티스 관절염 또는 크론병이 있는 257명의 여성과 아달리무맙을 투여받지않은 류마티스 관절염 또는 크론병이 있는 120명의 여성이 등록되었다.
일차 평가변수는 주요출생결함의 발생률이었다. 주요 출생결함이 있는 한 명이상의 생존 신생아 출산으로 종료된 임신율은 아달리무맙을 투여받은 여성에서 6/69(8.7%), 치료를 받지 않은 류마티스관절염 여성에서 5/74 (6.8%) (보정하지 않은 odd ratio 1.31, 95% 신뢰구간 0.38 ‑4.52)이었고, 아달리무맙을 투여받은 크론병 16/152 (10.5%), 치료를 받지 않은 크론병 여성에서 3/32 (9.4%)(보정하지 않은 odd ratio 1.14, 95% 신뢰구간 0.31 ‑4.16)이었다. 류마티스관절염과 크론병의 베이스라인 차이를 보정한 odd ratio는 1.10 (95% 신뢰구간 0.45 ‑2.73)이었다.
이차 평가변수인 경미한 출생결함, 조기분만, 저체중, 심각한 또는 기회 감염에서 유의한 차이가 관찰되지 않았다 (보정한 odd ratio 0.84, 95% 신뢰구간 0.34, 2.05). 사산이나 악성 종양은 보고되지 않았다.
이 등록연구는 적은 표본 크기와 무작위연구 설계가 아니라는 방법론적인 한계로 데이터 해석에 영향을 줄 수 있다.
아달리무맙은 임신 중 아달리무맙으로 치료받은 여성에서 태반을 통과하여 태아의 혈청으로 들어간다. 결과적으로 이 유아들은 감염의 위험이 증가될 수 있다. 자궁에서 아달리무맙에 노출된 유아의 경우, 임신 중 이 약의 마지막 투여 후 5개월 동안 생백신 투여는 권장되지 않는다.
2) 출판된 문헌의 제한된 정보에서 모유로 분비되는 아달리무맙의 농도는 매우 낮으며, 인간의 모유 내 아달리무맙 농도는 모체 혈청 수치의 0.1%에서1% 정도이다. 경구로 섭취한 면역글로불린 G 단백질은 장내에서 단백질 분해되고 생체이용률이 낮으므로, 모유수유하는소아에 대한 아달리무맙의 전신 영향 가능성은 거의 없을 것이다. 모유수유의 발달 및 건강상 이익은 아달리무맙에 대한 수유부의 임상적 필요성과 아달리무맙 또는 수유부의 기존 상태로 인한 소아에 대한 잠재적인 유해성과 함께 고려해야 한다.
7. 소아에 대한 투여
이 약은 소아 환자에 대해 연구되지 않았으므로 추가적인 자료를 얻을 때까지 18세 미만 소아에 대한 투여는 권장되지 않는다[소아 특발성 관절염(다관절형 소아 특발성 관절염, 골부착부위염 관련 관절염), 크론병(6 ‑ 17세) 및 판상 건선 제외].
8. 고령자에 대한 투여
이 약의 용량 조절이 요구되지 않는다. 이 약의 임상시험에 참가한 환자 중 65세 이상이 9.4%이고 75세 이상이 약 2.0%였으며 이들 환자군과 더 젊은 환자군 사이에 전반적인 유효성의 차이는 관찰되지 않았다. 이 환자군에서 용량조절은 필요하지 않다.
9. 과량투여시의 처치
임상시험 중 용량제한독성은 관찰되지 않았다. 평가된 최고 용량은 10 mg/kg의 수회 정맥투여였다. 과량투여의 경우 이상사례의 증상이나 징후 또는 효과를 모니터링하고 적절한 대증적 처치를 한다.
10. 적용상의 주의사항
1) 주사방법에 대한 교육을 받은 후 환자가 자가주사할 경우, 자가주사 부위는 대퇴부 또는 복부를 포함하며 주사부위는 교대로 바꿔야 한다. 새로운 주사부위는 앞서 주사한 부위에서 최소 3 cm 정도 떨어져서 주사한다.
2) 새로운 주사부위가 약하거나, 멍들거나, 충혈되거나 딱딱한 경우에는 투여해서는 안 된다. 투여 전 변색 및 이물질에 대해 육안으로 관찰한다. 주사액은 무색투명해야 하며, 주사액이 변색 또는 불투명하거나 이물이 있을 경우 투여하지 않는다. 펜형 제품의 경우 몸체에 있는 창(window)을 통해 확인한다. 주사 전 제품을 흔들거나 떨어뜨리지 않는다.
회색 뚜껑 및 자주색 뚜껑을 주사 직전에 제거한다.
3) 프리필드시린지
주사부위를 동봉된 알콜솜으로 닦는다. 주사바늘 덮개를 벗기고, 주사부위를 부드럽게 잡아 고정한 후 피부와 45도 각도로 주사한다.
4) 펜(pen) 형
⓵ 주사부위를 동봉된 알콤솜으로 닦는다. 회색 뚜껑 및 자주색 뚜껑을 주사 직전에 제거한다. 펜의 회색부분을 한손으로 잡는다. 펜의 회색 뚜껑(1) 및 자주색 뚜껑(2)에 손이 닿지 않도록 펜의 가운데를 손으로 잡는다. 회색 뚜껑(1)이 위로 향하도록 잡는다.
다른 손으로 회색 뚜껑(1)을 즉시 당겨서 제거한다. 작은 회색 주사바늘 커버와 덮개가 함께 제거되었는지 확인하며, 뚜껑을 다시 씌우면 안 된다. 주사침에서 몇 방울의 액이 나오는 것은 정상이다.
⓶ 펜의 자주색 뚜껑(2)을 잡아당겨 벗긴 후 버린다. 자주색 작동 버튼이 노출되어 펜을 사용할 준비가 된다. 약물이 방출될 수 있으므로, 적절한 주사위치를 잡기 전까지 자주색 작동 버튼을 누르거나 뚜껑을 다시 씌우면 안 된다.
⓷ 한 손으로 주사부위의 깨끗한 피부를 아래 그림처럼 부드럽게 잡아 고정시킨다.
⓸ 펜의 흰색 끝부분을 창이 보이도록 하여 피부와 직각(90도)이 되도록 놓는다. 창으로 보여 지는 몇 방울의 기포는 정상이다.
⓹ 펜의 몸체를 잡고 주사부위에 가볍게 눌러 주사준비를 한 다음 집게손가락 또는 엄지손가락으로 주사기 윗부분의 자주색 버튼을 누른다. 바늘이 나올 때 나는 ‘딸깍’ 소리가 들리고, 바늘에 찔리는 따끔함이 느껴진다.
➅ 약 10초간 유지하여 완전히 주사되도록 한다.
⑦ 주사 동안 노란색 표시가 펜의 창내로 이동하며, 노란색 표시의 이동이 멈추면 주사가 끝난 것이다. 노란색 표시는 펜의 주사막대의 일부분이다. 노란색 표시가 창에 나타나지 않는다면, 주사막대가 적절히 이동하지 않은 것이고, 주사가 완전히 이루어지지 않은 것이다.
5) 이 약의 주사기에 다른 약물을 혼합해서는 안 된다. 사용하지 않고 남은 부분은 버린다. 일회용이며 재사용하지 않는다. 어린이의 손이 닿지 않는 곳에 보관한다.
11. 저장상의 주의사항
1) 이 약은 동결을 피하여 냉장 보관(2 ‑ 8℃)한다. 사용 전까지 포장을 뜯지 않고 용기 상자 내부에 보관한다.
2) 이 약은 냉장조건을 벗어난 경우 1회에 한하여 25℃이하에서 최대 14일간 보관할 수 있으며, 빛을 피하여 보관하여야 한다. 냉장조건을 벗어난 제품은 다시 냉장보관해서는 안되며, 14일 이내 사용하거나 폐기하여야 한다.
12. 기타
이 약은 류마티스 관절염 환자에 대한 한 건의 이중 눈가림 후 공개연장 임상시험에서 최대 60개월까지 투여되었다. 60개월까지의 투여기간 동안 류마티스 관절염의 증상과 징후를 감소시키는 효과가 유지되었고 관절손상 진행속도 감소와 신체활동기능 향상 효과가 유지되었으며, 전반적으로 안전하고 내약성이 좋았다.
13. 전문가를 위한 정보
1) 임상약리학적 정보
⓵ 일반 정보
아달리무맙은 TNF에 특이적으로 결합하여 TNF와 p55 및 p75 세포 표면 TNF 수용체의 상호작용을 차단함으로써 TNF의 생물학적 기능을 중화시킨다. TNF는 자연적으로 발생하는 사이토카인으로서 정상적인 염증 및 면역 반응에 관여한다. 또한 아달리무맙은 백혈구 이동(ELAM ‑1, VCAM ‑1 및 ICAM ‑1, IC50 1 ‑2 × 10 ‑<SUP>10</SUP>M)의 원인이 되는 부착 분자의 수치 변화와 같이 TNF에 의해 유도되거나 조절되는 생물학적 반응을 조절한다.
⓶ 약력학적 정보
이 약을 투여한 후 류마티스 관절염 환자에서 베이스라인과 비교하여 염증의 급성 단계 반응물질(C ‑반응성 단백질[C ‑reactive protein; CRP]과 적혈구 침강속도[erythrocyte sedimentation rate; ESR] 과 혈중 사이토카인[IL ‑6]) 수치가 급격히 감소하였다. 또한 소아 특발성 관절염, 크론병, 궤양성 대장염 및 화농성 한선염 환자에서 CRP 수치 감소뿐만 아니라 크론병 환자의 대장에서 인간 백혈구 항원(human leukocyte antigen; HLA ‑DR) 및 골수세포형과산화효소(myeloperoxidase; MPO)와 같은 염증 표지자와 TNF가 유의하게 감소하였다. 연골 손상의 원인이 되는 조직 리모델링을 일으키는matrix metalloproteinase(MMP ‑1 및 MMP ‑3)의 혈중 수치 또한 이 약의 투여 후 감소하였다. 류마티스 관절염, 건선성 관절염, 강직성 척추염 환자는 경증부터 중등도의 빈혈 및 림프구 수 감소뿐 아니라 호중구와 혈소판 수 증가를 종종 경험한다. 이 약을 투여 받은 환자는 대개 만성 염증의 혈액학적 징후에서 개선을 경험하였다.
그림 1: 농도 ‑효과 연관성
⓷ 약동학적 정보
흡수
건강한 성인 대상자 59명에게 아달리무맙 40 mg을 단회 피하 투여한 아달리무맙의 흡수 및 분포는 느리며 투여 후 약 5일 뒤에 평균 최대 혈중 농도에 도달하였다. 3건의 시험에서 40 mg 단회 피하 투여 후 추정한 아달리무맙의 절대 생체이용률은 평균 64%였다.
분포 및 배설
아달리무맙의 단일 투여 약동학은 0.25 ‑10 mg/kg 범위의 정맥 내 투여에 대한 여러 시험에서 확인되었다. 4.7 ‑6.0 L 범위의 분포 용적(distribution volume; Vss)은 아달리무맙이 혈관과 혈관 외액 사이에 거의 비슷하게 분포함을 나타내었다. 아달리무맙은 일반적으로 12 mL/h 이하의 청소율로 천천히 배설된다. 평균 최종 단계 반감기는 약 2주였으며 범위는 10 ‑20일이었다. 청소율 및 반감기는 시험된 투여량 범위에서 상대적으로 변화가 없었고 최종 반감기는 IV 투여 및 SC 투여에서 비슷하였다. 여러 류마티스 관절염환자 활액에서의 혈중 아달리무맙 농도 범위는 31 ‑96%였다.
정상상태(steady ‑state)에서의 약동학
류마티스 관절염 환자에게 격주로 아달리무맙 40 mg을 SC 투여한 후의 반감기를 근거로 아달리무맙의 축적을 예측할 수 있었으며 평균 정상상태 최저 농도는 각각 약 5 mcg/mL(메토트렉세이트[MTX] 병용 투여하지 않은 경우) 및 8 ‑9 mcg/mL (MTX 병용 투여한 경우)였다. 정상상태 혈중 아달리무맙 최저 수치는 격주 및 매주 SC 투여한 20, 40 및 80 mg의 투여량에 따라 거의 비례적으로 증가하였다. 2년 이상의 장기간 투여 시험에서 시간에 따른 청소율의 변화는 확인되지 않았다.
건선 환자의 경우 메토트렉세이트의 병용 투여 없이 아달리무맙 40 mg 격주 투여 동안 평균 정상상태 최저 농도는 5 mcg/mL였다.
0주차에 아달리무맙 160mg을 투여 받은 후 2주차에 80 mg을 투여 받은 화농성 한선염 환자의 혈중 아달리무맙 최저 농도는 2주차와 4주차에 약 7 ‑8 mcg/mL에 도달하였다. 12주차부터 36주차까지의 평균 정상상태 최저 농도는 매주 아달리무맙 40 mg 투여 동안 약 8 ‑10 mcg/mL였다.
포도막염 환자의 경우 0주차에 도입용량인 아달리무맙 80 mg을 투여하고 1주차부터 격주마다 아달리무맙 40 mg을 투여했을 때 평균 정상상태 농도는 약 8 ‑10 mcg/mL였다.
집단 약동학과 약동학/약력학 모델링 및 시뮬레이션 결과, 매주 40 mg의 아달리무맙 투여와 비교하여 격주 80 mg 투여한 환자의 아달리무맙 노출 및 효능이 유사한 것으로 예측하였다(성인 류마티스 관절염, 화농성 한선염, 궤양성 대장염, 크론병 또는 건선환자 및 40kg 이상의 소아 크론병 환자).
1,200명 이상 환자 데이터의 집단 약동학 분석을 통해 메토트렉세이트의 병용 투여가 아달리무맙의 겉보기 청소율(apparent clearance; CL/F)에 본질적인 영향을 미친다는 것이 확인되었다(‘5. 상호작용’ 참조). 예상한 바와 같이 체중 증가 및 항 ‑아달리무맙 항체의 존재 하에 아달리무맙의 겉보기 청소율이 높아지는 경향이 있었다.
보다 중요하지 않은 다른 요인들도 확인하였다. 권장된 투여량보다 낮은 투여량을 받은 환자, 높은 류마티스인자를 가진 환자 또는 CRP 농도가 높은 환자의 겉보기 청소율은 높을 것으로 예측되었다. 이러한 요인은 임상적으로 중요할 가능성이 낮다.
비방사선학적 축성 척추관절염 환자에게 격주마다 아달리무맙 40 mg을 피하투여한 후 68주차의 평균(±SD) 최저 정상상태 농도는 8.0 ± 4.6 μg/mL였다.
크론병 환자의 경우, 0주차에 유도용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 2주차와 4주차의 평균 혈중 아달리무맙 최저 수치는 약 12 mcg/mL였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 크론병 환자에서 24주차와 56주차의 평균 정상상태 최저 수치는 약 7 mcg/mL로 관찰되었다.
궤양성 대장염 환자의 경우, 0주차에 유도용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 유도 기간 동안의 혈중 아달리무맙 최저 농도는 약 12 mcg/mL에 도달하였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 궤양성 대장염 환자에서의 평균 정상상태 최저 수치는 약 8 mcg/mL로 관찰되었다.
베체트병이 있는 일본 환자의 경우, 0주차에 도입용량인 아달리무맙 160 mg을 투여하고 2주차에 아달리무맙 80 mg을 투여했을 때 유도 기간 동안의 혈중 아달리무맙 최저 농도는 약 13 mcg/mL에 도달하였다. 격주마다 아달리무맙 40 mg의 유지용량을 투여 받은 장내 베체트병이 있는 일본 환자에서의 평균 정상상태 최저 수치는 약 9 mcg/mL로 관찰되었다.
특수 집단
특수한 집단에서의 약동학은 집단 약동학 분석을 사용하여 조사하였다.
고령 환자
연령은 아달리무맙의 겉보기 청소율에 대해 최소한의 영향을 미치는 것으로 나타났다. 집단 분석에서 40 ‑ 65세 환자(n=850) 및 ≧ 65세 환자(n=287)의 평균 체중을 보정한 청소율은 각각 0.33 및 0.30mL/h/kg이었다.
소아 환자
다관절형 소아 특발성 관절염이 있는 4 ‑ 17세의 환자에게 격주마다 아달리무맙 24 mg/m2(최대 40 mg)를 피하투여한 후 평균 최저 정상상태(20 ‑48주차에 측정한 수치) 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여했을 때 5.6 ± 5.6 μg/mL (102% CV), 메토트렉세이트를 병용 투여하지 않았을 때 10.9 ± 5.2 μg/mL (47.7% CV)였다. 20 mg 아달리무맙을 격주마다 피하투여한 30 kg인 환자에서 메토트렉세이트를 병용 투여하거나 병용 투여하지 않았을 때 평균 정상상태 혈중 아달리무맙 농도는 각각 6.8 μg/mL 및 10.9 μg/mL였다. 40 mg 아달리무맙을 격주마다 피하투여한 ≧30 kg인 환자에서 메토트렉세이트를 병용 투여하거나 병용 투여하지 않았을 때 평균 정상상태 혈중 아달리무맙 농도는 각각 6.6 μg/mL 및 8.1 μg/mL였다. 다관절형 소아 특발성 관절염이 있는 2세이상 4세미만 또는 ≧4세의 15 kg인 환자에게 아달리무맙 24 mg/m2를 투여했을 때의 평균 최저 정상상태 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여하지 않은 경우 6.0 ± 6.1 μg/mL (101% CV), 메토트렉세이트를 병용 투여한 경우 7.9 ± 5.6 μg/mL (71.2% CV)였다.
골부착부염 관련 관절염 환자에게 24 mg/m2(최대 40 mg)를 격주마다 피하투여한 후 평균 최저 정상상태(24주차에 측정한 수치) 혈중 아달리무맙 농도는 아달리무맙과 메토트렉세이트를 병용 투여하지 않은 경우 8.8 ± 6.6 μg/mL, 메토트렉세이트를 병용 투여한 경우 11.8 ± 4.3 μg/mL였다.
중등증부터 중증의 활동성 크론병 소아 환자에서의 공개 연장 시험 아달리무맙 유도용량은 0주차와 2주차에 체중 절단점(cut ‑off)인 40 kg에 따라 각각 160/80 mg 또는 80/40 mg이었다. 4주차에 환자는 체중에 따라 표준용량(격주 40/20 mg) 또는 저용량(격주 20/10 mg) 유지 투여군에 1:1의 비율로 무작위 배정되었다. 4주차에 도달한 평균(±SD) 혈중 아달리무맙 최저 농도는 ≧40 kg인 환자(160/80mg)의 경우 15.7 ± 6.6 μg/mL, 40 kg인 환자(80/40 mg)의 경우 10.6 ± 6.1 μg/mL였다.
무작위 배정된 치료를 지속한 환자에서 52주차의 평균(±SD) 아달리무맙 최저 농도는 표준용량 투여군의 경우 9.5 ± 5.6 μg/mL, 저용량 투여군의 경우 3.5 ± 2.2 μg/mL였다. 52주 동안 아달리무맙 격주 투여를 지속한 환자에서 평균 최저 농도는 유지되었다. 격주에서 매주 투여 요법으로 투여량을 증량한 환자의 경우 52주차의 평균(±SD) 아달리무맙 혈중 농도는 각각 15.3 ± 11.4 μg/mL(매주 40/20 mg) 및 6.7 ± 3.5 μg/mL(매주 20/10 mg)였다.
만성 판상형 건선이 있는 소아 환자에게 격주로 아달리무맙 0.8 mg/kg(최대 40 mg)을 피하투여한 후 평균 ±SD 정상상태 아달리무맙 최저 농도는 약 7.4 ± 5.8 μg/mL (79% CV)였다.
성별
환자의 체중을 보정한 후 성별과 관련된 약동학 차이는 관찰되지 않았다.
인종
인종간 면역글로불린 청소율의 차이는 예상되지 않는다. 코카시안이 아닌 인종의 제한된 데이터로부터, 아달리무맙에 대한 중요한 약동학적 차이는 관찰되지 않았다.
간장애 및 신장애 환자
간 또는 신장 기능 장애가 있는 환자에서 유효한 약동학 데이터가 없다.
질병 상태
건강한 지원자와 류마티스 관절염 환자는 유사한 아달리무맙 약동학을 보였다.
2) 임상시험 정보
⓵ 성인
류마티스 관절염
모든 류마티스 관절염 임상시험에서 3,000명 이상의 환자를 대상으로 이 약을 평가하였다. 이 약의 안전성 및 유효성은 5건의 무작위배정, 이중눈가림 된 시험에서 평가하였다. 일부 환자는 60개월 이상의 기간 동안 이 약을 투여 받았고, 일부는 최대 120개월의 기간 동안 이 약을 투여 받았다. 이 약 40 mg/0.8 mL과 비교한 이 약 40 mg/0.4 mL에 대한 투여 부위 통증은 2건의 무작위 배정, 활성대조, 단일 눈가림, 두 기간 교차시험으로 평가하였다.
RA 시험 I (DE009)은 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 271명을 대상으로 평가하였으며, 이 환자들은 1 ‑4개의 DMARDs(disease ‑modifying anti ‑rheumatic drug),(예: 히드록시클로로퀸, 경구 또는 주입 가능한 금, 아자티오프린, D ‑페니실라민, 설파살라진) 치료에 실패 이력이 있고 매주 12.5 ‑25 mg (메토트렉세이트 내약성이 없는 경우 10 mg)의 메토트렉세이트에 대해 효능이 불충분하며 메토트렉세이트 투여량을 매주 10 ‑25 mg으로 일정하게 유지하였다. 환자는 6개 이상의 종창 관절 및 9개 이상의 압통 관절이 있었으며 ACR 기준에 따라 류마티스관절염로 진단되었다. 이들은 24주 동안 격주마다 20, 40 또는 80 mg의 이 약이나 위약을 투여 받았다.
RA 시험 II (DE011)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 544명을 대상으로 평가하였으며, 이들은 최소 하나의 DMARD(예: 메토트렉세이트, 설파살라진, 히드록시클로로퀸, 경구 또는 주입 가능한 금, D ‑페니실라민, 아자티오프린) 치료에 실패한 이력이 있었다. 환자는 10개 이상의 종창 관절 및 12개 이상의 압통 관절이 있었으며 ACR 기준에 따라서도 진단되었다. 이들은 26주 동안 이 약 20 mg 또는 40 mg을 위약과 격주로 번갈아 피하 투여 받거나 매주 피하 투여 받았다. 위약은 동일한 기간 동안 매주 투여 받았다.
RA 시험 III (DE019)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 619명을 대상으로 평가하였으며, 매주 12.5 ‑25 mg (메토트렉세이트 내약성이 없는 경우 10 mg)의 메토트렉세이트에 대해 효능이 불충분하며 메토트렉세이트 투여량을 매주 12.5 ‑25 mg으로 일정하게 유지하였다. RA 시험 I과는 다르게, RA 시험 III의 환자는 메토트렉세이트 외의 DMARD 치료 실패 이력이 필요하지 않았다. 환자는 6개 이상의 종창 관절 및 9개 이상의 압통 관절이 있었으며 ACR 기준에 따라 류마티스 관절염으로 진단되었다. 이 시험에는 3개의 군이 있었다. 첫 번째 군은 52주 동안 매주 위약을 투여 받았다. 두 번째 군은 52주 동안 매주 이 약 20 mg 을 투여 받았다. 세 번째 군은 이 약 40 mg과 위약을 격주로 번갈아 투여 받았다. 첫 52주 동안 투여를 완료한 후 457명의 환자는 최대 60개월 동안 격주로 이 약 40 mg/메토트렉세이트를 투여 받는 공개 연장 시험에 등록되었다. 첫 52주 동안 투여를 완료한 후 457명의 환자는 최대 10년 동안 격주로 이 약 40mg /메토트렉세이트를 투여 받는 공개 연장 시험에 등록되었다.
RA 시험 IV (DE031)는 중등증에서 중증의 활동성 류마티스 관절염이 있는 18세 이상의 환자 636명을 평가하였다. 이 환자들은 최소 3개월 동안 류마티스 관절염 진단에 대한 ACR 기준을 충족했으며 최소 6개의 종창 관절 및 9개의 압통 관절을 가지고 있었다. 최소한 28일 동안 치료가 안정적이었을 경우, DMARD ‑I 또는 기존의 류마티스 치료를 유지하는 것이 허용되었다. 환자는 24주 동안 이 약 격주 40 mg투여군 또는 위약군에 무작위 배정되었다.
RA 시험 V (DE013)는 중등증에서 중증의 활동성 초기 류마티스 관절염(평균 질병 기간이 9개월 미만)이 있는 메토트렉세이트 투여 이력이 없는 성인 환자 799명을 대상으로 평가하였다. 이 시험에서는 104주 동안 류마티스 관절염에서 류마티스 관절염의 징후 및 증상을 감소시키고 관절 손상의 진행 속도를 줄이는 데 대한 이 약 40 mg 격주/메토트렉세이트 병용 치료, 이 약 40 mg 격주 단일 치료 및 메토트렉세이트 단일 치료의 효능을 평가하였다. 첫 104주 동안 투여를 완료한 후 497명의 환자는 최대 10년 동안 이 약 격주로 40 mg을 투여 받는 공개 연장 시험에 등록되었다.
RA 시험 VI 및 VII는 각각 중등증에서 중증의 활동성 류마티스 관절염이 있는 ≧ 18세 환자 60명을 대상으로 평가하였다. 등록된 환자는 당시 이 약 40 mg/0.8 mL를 투여 받는 중이며 평균 투여 부위 통증이 최소 3cm (0 ‑10cm VAS)인 환자, 또는 이 약 40 mg/0.8 mL를 투여 받기 시작한 생물학적 치료 이력이 없는 환자였다. 환자는 이 약 40 mg/0.8 mL 또는 이 약 40 mg/0.4 mL 단일 투여군으로 무작위 배정된 후 다음 파트에 교차 투여된다.
RA 시험 I ‑V의 결과는 ACR 반응 기준을 사용하여 류마티스 관절염에서 개선을 보인 환자 비율로 표현하였다. RA 시험 I, II 및 III의 일차 평가변수와 RA 시험 IV의 이차 평가변수는 24주 또는 26주차에 ACR 20 반응에 도달한 환자의 비율이었다. RA 시험 V의 일차 평가변수는 52주차에 ACR 50 반응에 도달한 환자 비율이었다. RA 시험 III 및 V의 52주차에서 추가적인 일차 평가변수는 질병 진행의 지연이었다(엑스레이 결과로 확인). RA 시험 III 또한 삶의 질 변화를 일차 평가변수로 설정하였다. RA 시험 VI 및 VII의 일차 평가변수는 0 ‑10 cm VAS로 측정한 투여 직후의 투여 부위 통증이었다.
임상 반응
RA 시험 I, II 및 III
이 약을 투여 받은 환자 중 ACR 20, 50 및 70 반응에 도달한 환자 비율은 3건의 시험 모두에서 일관적이었다. 3건의 시험 결과는 표 3에 요약한다.
표 3: 위약대조 시험에서의 ACR 반응(환자 비율)
|
반응 |
RA 시험 Ia* |
RA 시험 IIa* |
RA 시험 IIIa* | |||
|
|
위약/MTX |
이 약b/MTX |
위약/MTX |
이 약b/MTX |
위약/MTX |
이 약b/MTX |
|
|
n=60 |
n=63 |
n=110 |
*n=113 |
n=200 |
n=207 |
|
ACR 20 |
|
|
|
|
|
|
|
6개월 |
13.3% |
65.1% |
19.1% |
46.0% |
29.5% |
63.3% |
|
12개월 |
NA |
NA |
NA |
NA |
24.0% |
58.9% |
|
ACR 50 |
|
|
|
|
|
|
|
6개월 |
6.7% |
52.4% |
8.2% |
22.1% |
9.5% |
39.1% |
|
12개월 |
NA |
NA |
NA |
NA |
9.5% |
41.5% |
|
ACR 70 |
|
|
|
|
|
|
|
6개월 |
3.3% |
23.8% |
1.8% |
12.4% |
2.5% |
20.8% |
|
12개월 |
NA |
NA |
NA |
NA |
4.5% |
23.2% |
a RA 시험 I 24주차, RA 시험 II 26주차, RA 시험 III 24주차 및 52주차
b 40mg 이 약 격주 투여
* p0.01, ACR 20, 50, 70에 대한 모든 시점에서 이 약 vs. 위약
RA 시험 II에서 매주 이 약 40 mg을 투여 받은 환자 또한 6개월차에 통계적으로 유의하게 ACR 20, 50 및 70 반응률에 각각 53.4%, 35.0% 및 18.4% 도달하였다.
RA 시험 III에 대한 ACR 반응 기준의 구성요소 결과는 표 4에 제시되었다. 아래에 제시한 결과는 일반적으로 수행된 각 시험을 대표하는 것이다.
ACR 반응률 및 모든 ACR 반응 기준의 개선은 104주차까지 유지되었다. RA 시험 III에서 2년 동안 이 약 투여군의 24%가 6개월 동안 ACR 70 반응을 유지한 것으로 정의되는 주요 임상 반응에 도달하였다. ACR 반응은 RA 시험 III의 공개 연장 시험에서 이 약 치료를 지속한 환자 중 비슷한 비율의 환자에서 최대 60개월 동안 유지되었다.
RA 시험 III의 공개 연장에서 ACR 반응자 중 대다수의 환자가 최대 10년간의 추적조사 시 반응을 유지했다. 격주 이 약 40 mg 투여군으로 무작위 배정된 환자 207명 중 114명은 5년 동안 격주로 이 약 40 mg을 지속적으로 투여 받았다. 이들 중 86명(75.4%)은 ACR 20 반응, 72명(63.2%)은 ACR 50 반응, 41명(36%)은 ACR 70 반응에 도달하였다. 207명의 환자 중 81명은 10년 동안 이 약 40 mg을 지속적으로 투여 받았다. 이들 중 64명(79.0%)은 ACR 20 반응, 56명(69.1%)은 ACR 50 반응, 43명(53.1%)은 ACR 70 반응에 도달하였다.
표 4: RA 시험 III에서의 ACR 반응 구성요소
|
|
베이스라인 |
24주차 |
52주차 |
베이스라인 |
24주차 |
52주차 |
|
매개변수(중앙값) |
위약/MTX (N=200) |
이 약a/MTX (N=207) | ||||
|
압통 관절 수(0-68) |
26.0 |
15.0 |
15.0 |
24.0 |
8.0* |
6.0* |
|
종창 관절 수(0-66) |
17.0 |
11.0 |
11.0 |
18.0 |
5.0* |
4.0* |
|
의사의 전반적 질병 활성도 평가b |
63.0 |
35.0 |
38.0 |
65.0 |
20.0* |
16.0* |
|
환자의 전반적 질병 활성도 평가b |
53.5 |
39.0 |
43.0 |
52.0 |
20.0* |
18.0* |
|
통증b |
59.5 |
38.0 |
46.0 |
58.0 |
21.0* |
19.0* |
|
장애 척도(HAQ)c |
1.50 |
1.25 |
1.25 |
1.50 |
0.75* |
0.75* |
|
CRP (mg/L) |
10.0 |
9.0 |
9.0 |
10.0 |
4.0* |
4.0* |
|
a 40mg 이 약 격주 투여 b 시각적 아날로그 척도. 0점 = 최고, 100점 = 최악 c 건강 평가 설문조사의 장애 척도. 0점 = 최고, 3점 = 최악. 옷 입기/치장하기, 일어나기, 먹기, 걷기, 뻗기, 잡기, 위생 유지 및 일상 활동 유지 등을 수행할 수 있는 환자의 능력 측정 * p <0.001, 이 약 vs. 위약, 베이스라인으로부터 평균 변화에 기초함 | ||||||
RA 시험 III에서 24주차에 ACR 20 반응에 도달한 환자의 84.7%는 52주차에도 반응을 유지하였다. 다음의 그림은 시험 III 및 II에서이 약에 대한 ACR 20 반응의 지속성을 나타낸다.
RA 시험 IV
이 약과 함께 표준치료를 투여 받은 환자의 ACR 20 반응은 위약과 함께 표준치료를 투여 받은 환자에 비해 통계적으로 유의하게 우수하였다(p0.001). 이 약과 다른 DMARD의 병용 투여와 관련된 고유의 이상사례는 관찰되지 않았다.
RA 시험 I ‑IV에서 이 약을 투여 받은 환자는 위약을 투여 받은 환자보다 더 빠르고 자주 ACR 20, 50 및 70 반응에 도달하였다. RA 시험 I에서 1주차(첫 시험 방문)의 ACR 20 반응에 대해 이 약을 투여 받은 환자(26.0%)와 위약을 투여 받은 환자(5.0%) 사이에 통계적으로 유의한 차이가 있었다. ACR 20 반응에 대한 통계적으로 유의한 차이는 RA 시험 II, III 및 IV의 2주차(첫 시험 방문)에 이 약을 투여 받은 환자(각각 36.4%, 29.1% 및 33.7%)와 위약을 투여 받은 환자(각각 7.3%, 13.0% 및 8.6%) 사이에서도 확인되었다. 첫 ACR 50 및 70 반응까지의 시간은 4건의 시험 모두에서 유사한 패턴으로 나타났다.
메토트렉세이트를 병용 투여하지 않은 일부 환자에서 매주 40 mg으로 이 약의 투여 빈도를 증가시킴에 따라 추가 이익이 유도될 수 있다. 이는 불완전한 반응을 보인 환자의 투여 빈도를 격주 40 mg에서 매주 40mg으로 증가시킨 장기간 공개 연장 시험에서 확인되었다.
RA 시험 V
메토트렉세이트의 투여 이력이 없는 초기 류마티스 관절염 환자를 대상으로 한 RA 시험 V에서, 이 약과 메토트렉세이트의 병용 치료는 52주차에 메토트렉세이트 단일 치료 및 이 약 단일 치료와 비교하여 더 빠르고 유의하게 큰 ACR 반응을 유발했으며 이 반응은 104주까지 지속되었다(표 5참조).
52주차에 ACR 반응 기준의 모든 개별적인 구성요소는 이 약/메토트렉세이트 치료로 인해 개선되었으며 개선은 104주까지 유지되었다.
2년간의 시험 동안, 이 약/메토트렉세이트 병용 치료를 받은 환자 중 48.5%가 주요 임상 반응(6개월 연속 ACR 70)에 도달했으며, 이에 비해 메토트렉세이트 단일 치료를 받은 환자는 27.2% (p0.001), 이 약 단일 치료를 받은 환자는 24.5% (p0.001)가 주요 임상 반응에 도달하였다.
표 5: RA 시험 V에서의 ACR 반응(환자 비율)
|
반응 |
MTXb N=257 |
이 약c N=274 |
이 약/MTX N=268 |
|
ACR 20 | |||
|
52주차 |
62.6% |
54.4% |
72.8% |
|
104주차 |
56.0% |
49.3% |
69.4% |
|
ACR 50 | |||
|
52주차 |
45.9% |
41.2% |
61.6% |
|
104주차 |
42.8% |
36.9% |
59.0% |
|
ACR 70 | |||
|
52주차 |
27.2% |
25.9% |
45.5% |
|
104주차 |
28.4% |
28.1% |
46.6% |
|
주요 임상 반응a | |||
|
104주차 |
27.2% |
24.5% |
48.5% |
a 주요 임상 반응은 6개월 연속 ACR 70 반응에 도달한 것으로 정의한다.
b p0.05, ACR 20에 대한 이 약/MTX vs. MTX
p0.001, ACR 50, 70 및 주요 임상 반응에 대한 이 약/MTX vs. MTX
c p0.001, 이 약/MTX vs. 이 약
RA 시험 V의 공개 연장시험에서 ACR 반응률은 최대 10년간의 추적조사 시에 유지되었다. 이 약 격주 40 mg 투여군으로 무작위 배정된 환자 542명 중 170명은 10년 동안 이 약 격주 40 mg 투여를 지속하였다. 이들 중 154명(90.6%)은 ACR 20 반응, 127명(74.7%)은 ACR 50 반응, 102명(60.0%)은 ACR 70 반응에 도달하였다. 52주차에 이 약/메토트렉세이트 병용 치료를 받은 환자 중 42.9%가 임상적 관해(질병 활성도 점수[Disease Activity Score; DAS28] ‑CRP 2.6)에 도달하였고, 이에 비해 메토트렉세이트 단일 치료를 받은 환자는 20.6%, 이 약 단일 치료를 받은 환자는 23.4%가 임상적 관해에 도달하였다. 이 약/메토트렉세이트 병용 치료는 최근에 중등증부터 중증의 류마티스 관절염을 진단 받은 환자가 낮은 질병 상태에 도달하는 데 있어 메토트렉세이트(p0.001) 및 이 약 단일 치료(p0.001)보다 통계적 및 임상적으로 우수하였다(표 6참조). 기존에 이 약 단일 치료 또는 이 약/메토트렉세이트 병용 치료로 무작위 배정되어 공개 연장 시험에 등록된 342명의 대상자 중 171명이 10년 동안의 이 약 치료를 완료하였다. 이들 중 109명(63.7%)이 10년차에 질병 관해를 보고하였다.
표 6: RA 시험 V에서의 DAS28 반응
|
DAS28 반응 |
MTX N=257 |
이 약 N=274 |
이 약/MTX N=268 |
|
52주차 | |||
|
베이스라인(평균) |
6.3 |
6.4 |
6.3 |
|
베이스라인으로부터 평균 변화(평균 ± SD) |
-2.8 ± 1.4a |
-2.8 ± 1.5b |
-3.6 ± 1.3 |
|
관해된 환자 비율(DAS28 <2.6) |
20.6%a |
23.4%b |
42.9% |
|
104주차 | |||
|
베이스라인(평균) |
6.3 |
6.3 |
6.3 |
|
베이스라인으로부터 평균 변화(평균 ± SD) |
-3.1 ± 1.4a |
-3.2 ± 1.4b |
-3.8 ± 1.3 |
a p0.001, 이 약/메토트렉세이트 vs. 메토트렉세이트
b p0.001, 이 약/메토트렉세이트 vs. 이 약
방사선학적 반응
RA 시험 III에서 이 약을 투여 받은 환자의 류마티스 관절염 평균 지속 기간은 약 11년이었으며 구조적 관절 손상은 방사선 촬영을 통해 평가하였고 수정된 전체 샤프 점수(modified total Sharp score; TSS) 및 구성요소, 미란(erosion) 점수 및 관절 간격 협착(joint space narrowing score; JSN)의 변화로 결과를 표시하였다. 손/손목 및 발의 앞부분의 방사선 사진은 베이스라인, 6개월 및 12개월에 판독하였다. 12개월 결과는 표 7에 제시한다. 6개월 때 TSS 및 미란 점수의 변화에서 통계적으로 유의한 차이가 관찰되었고 12개월 때 유지되었다. 52주차에 이 약/메토트렉세이트를 투여 받은 환자는 MTX만 단독 투여 받은 환자보다 방사선학적 진행이 더딘 것으로 확인되었다.
표 7: MTX를 병용 투여한 RA 시험 III에서 12개월간의 방사선학적 변화
|
|
위약 N=200 |
이 약a N=207 |
이 약a와 위약간 차이 |
p값 |
|
수정된 전체 샤프 점수의 변화(평균) |
2.7 |
0.1 |
-2.6 |
≤0.001b |
|
미란의 변화(평균) |
1.6 |
0.0 |
-1.6 |
≤0.001 |
|
새로운 미란 없음(환자 비율 %) |
46.2 |
62.9 |
16.7 |
≤0.001 |
|
JSN 점수의 변화(평균) |
1.0 |
0.1 |
-0.9 |
0.002 |
a 격주 40mg 투여
b ranked ANCOVA 분석에 기초함
RA 시험 III의 공개 연장 시험에서 특정 용량의 이 약을 투여 받은 기존의 환자 중 77%는 2년차에 방사선 촬영으로 평가하였다. TSS로 측정한 결과, 환자는 구조적 손상의 억제를 유지했다. 54%의 환자는 구조적 손상의 진행이 없었다(0점 이하의 TSS 변화로 정의). 격주로 이 약 40 mg을 기존에 투여 받은 환자 중 55%는 5년차에 방사선 촬영으로 평가하였다. 환자의 구조적 손상 억제는 지속적이었으며 50%는 구조적 손상의 진행이 없었다(0점 이하의 TSS 변화로 정의).
RA 시험 III의 공개 연장 시험에서 구조적 손상의 진행률 감소는 환자의 하위군에서 8 ‑10년 동안 유지되었다. 격주로 이 약 40 mg을 기존에 투여 받은 환자 207명 중 81명의 환자를 8년차에 방사선 촬영으로 평가하였다. 이들 중 48명의 환자는 구조적 손상의 진행이 없었다(베이스라인으로부터 mTss 변화가 0.5 이하). 격주로 이 약 40 mg을 기존에 투여 받은 환자 207명 중 79명의 환자를 10년차에 방사선 촬영으로 평가하였다. 이들 중 40명의 환자는 구조적 손상의 진행을 보이지 않았다(베이스라인으로부터 mTss 변화가 0.5 이하).
RA 시험 V에서의 구조적 관절 손상은 RA 시험 III과 동일하게 평가하였다. 방사선학적 진행의 억제(TSS, 미란 점수 및 JSN의 변화로 평가)는 52주차와 104주차 모두, 메토트렉세이트 또는 이 약 단일 투여군보다 이 약/메토트렉세이트 병용 투여군에서 높게 관찰되었다(표 8참조).
표 8: RA 시험 V에서 52주차의 방사선학적 평균 변화
|
|
MTXa N=257 (95% 신뢰구간) |
이 약b N=274 (95% 신뢰구간) |
이 약/MTX N=268 (95% 신뢰구간) |
|
전체 샤프 점수 |
5.7 (4.2-7.3) |
3.0 (1.7-4.3) |
1.3 (0.5-2.1) |
|
미란 점수 |
3.7 (2.7-4.7) |
1.7 (1.0-2.4) |
0.8 (0.4-1.2) |
|
JSN 점수 |
2.0 (1.2-2.8) |
1.3 (0.5-2.1) |
0.5 (0-1.0) |
a p0.001, 52주차 및 104주차에서 이 약/MTX vs. MTX
b p0.01, 52주차 이 약/MTX vs. 이 약
p0.001, 104주차 이 약/MTX vs. 이 약
52주 및 104주간의 투여 이후, 질병 진행이 없는(베이스라인으로부터 수정된 전체 샤프 점수의 변화 ≦0.5점) 환자의 비율은 이 약/메토트렉세이트 병용 치료군(각각 63.8% 및 61.2%)이 메토트렉세이트 단일 치료군(각각 37.4%, 33.5%, p0.001)과 이 약 단일 치료군(각각 50.7%, p0.002 및 44.5%, p0.001)에 비해 유의하게 높았다.
RA 시험 V의 공개 연장 시험에서 베이스라인으로부터 10년차까지 수정된 전체 샤프 점수의 평균 변화는 메토트렉세이트 단일 치료군, 이 약 단일 치료군 및 이 약/메토트렉세이트 병용 치료군으로 기존에 무작위 배정된 환자에서 각각 10.8점, 9.2점 및 3.9점이었다. 방사선학적 진행이 없는 환자의 비율은 각각 31.3%, 23.7% 및 36.7%였다.
삶의 질 및 신체적 기능
5건의 적절하고 잘 제어된 시험 모두에서 건강 평가 설문조사(Health Assessment Questionnaire; HAQ)의 장애 척도를 사용하여 건강 관련 삶의 질을 평가하였고, 이는 RA 시험 III에서 사전에 명시된 52주차의 일차 평가변수였다. 4건의 시험 모두에서 이 약의 모든 투여/계획은 베이스라인으로부터 6개월차까지 HAQ의 장애 척도에 대해 위약보다 통계적으로 유의하게 큰 개선을 보였다. RA 시험 III에서 베이스라인으로부터 52주차까지의 HAQ 평균(CI) 개선은 이 약/메토트렉세이트 환자의 경우 ‑0.60 ( ‑0.65, ‑0.55)이었고 위약/메토트렉세이트 환자(p0.001)의 경우 ‑0.25 ( ‑0.33, ‑0.17)였다. 이 약/메토트렉세이트를 투여 받은 환자 중 63%는 시험의 이중눈가림 부분에서 52주차에 0.5 이상의 HAQ 개선에 도달하였다. 이 환자들 중 82%는 104주차까지 개선이 유지되었으며 비슷한 비율의 환자가 공개 연장시험의 260주차(5년)까지 반응이 유지되었다. 신체적 기능이 개선되고 치료를 지속한 대부분의 환자는 공개 연장 시험의 520주차(10년)까지 개선이 유지되었다.
적절하게 잘 제어된 4건의 시험 모두에서 일반적인 건강 관련 삶의 질을 평가하기 위해 약식 건강 조사(SF ‑36) 또한 사용하였다. 4건의 시험 모두에서 이 약의 모든 투여/계획은 베이스라인으로부터 6개월차까지의 SF ‑36 신체적 구성요소 요약 점수에 대해 통계적으로 유의하게 큰 개선을 보였고 이는 RA 시험 III의 52주차에서도 유지되었다. SF ‑36의 평균 개선 또한 156주차(36개월)의 측정 종료 시점까지 유지되었다. 시험 II 및 IV의 SF ‑36 정신적 구성요소 요약 점수 또한 6개월차에 이 약이 위약보다 통계적으로 유의하게 높았다. 이 약을 40mg 격주투여한 4건의 시험 모두에서 베이스라인으로부터 6개월차까지 SF ‑36의 통증 및 활력 영역 점수는 위약과 비교하여 이 약을 투여 받은 환자에서 통계적으로 유의하게 큰 개선을 보였다. 이러한 결과는 분석한 3건의 시험 모두 6개월차에 피로가 통계적으로 유의하게 감소함을 나타낸 만성질환 치료의 기능적 평가(functional assessment of chronic illness therapy; FACIT) 점수로 뒷받침되었으며 RA 시험 III의 52주차에서도 유지되었다.
RA 시험 V에서 HAQ 장애 척도 및 SF ‑36의 신체적 구성요소는 52주차에 메토트렉세이트 단일 치료군 및 이 약 단일 치료군 모두와 비교하여 이 약/메토트렉세이트 병용 치료군에서 더욱 개선되었다(p0.001). 이 개선은 104주차까지 유지되었다. 공개 연장시험을 완료한 250명의 대상자에서 신체적 기능의 개선은 10년의 치료 동안 유지되었다.
투여 부위 통증
통합 교차 RA 시험 VI 및 VII의 경우 아달리무맙 40mg/0.8mL과 아달리무맙 40mg/0.4mL 사이에서 투여 직후 투여 부위 통증에 대해 통계적으로 유의한 차이가 관찰되었다(평균 VAS 3.7cm vs. 1.2cm, 0 ‑10cm 척도, p0.001). 이는 투여 부위 통증의 84% 중앙값 감소를 나타낸다.
건선성 관절염
2건의 무작위배정, 이중눈가림, 위약 대조 시험에서 건선성 관절염 환자 413명을 대상으로 이 약 안전성과 유효성을 평가하였다. PsA 시험 I (M02 ‑518)에는 중등증부터 중증의 활동성 건선성 관절염(>3개의 종창 관절 및 >3개의 압통 관절)이 있는 성인 환자 313명이 등록되었으며 이들은 다음의 유형 중 하나에서 NSAID 치료에 대해 부적절한 반응을 보였다: (1) 원위지절간관절(distal interphalangeal; DIP) 관련(N=23), (2) 다관절형 관절염(류마티스 결절이 없고 건선이 존재)(N=210), (3) 단절성 관절염(N=1), (4) 비대칭성 건선성 관절염(N=77), 또는 (5) 강직성 척추염 유사(N=2). 등록 시 MTX 치료(>1개월 동안 안정적 용량 ≦30 mg/주) 중이었던 환자(313명 중 158명)는 동일 용량으로 메토트렉세이트를 계속 투여 받을 수 있었다. 24주의 이중눈가림 기간 동안 격주로 이 약 40 mg 또는 위약을 투여 받았다. 12주간 수행한 PsA 시험 II (M02 ‑570)는 DMARD 치료에 부적절한 반응을 보인 100명의 환자를 대상으로 하였다. 두 시험의 완료 후 383명의 환자는 공개 연장시험에 등록되어 격주로 40 mg의 이 약을 투여 받았다.
ACR 및 PASI 반응
위약과 비교하여, 이 약의 치료는 질병 활성도 측정에서 개선을 보였다(표 9 및 10 참조). 이 약을 투여 받은 건선성 관절염 환자 중, 첫 방문 시점(2주차)에서 일부 환자의 임상적 반응이 명백하였다. 비록 단절성 관절염 및 강직성 척추염 유사 하위유형에는 극소수의 환자가 등록되었지만 PsA의 각 하위유형 환자에서도 비슷한 반응이 관찰되었다. 베이스라인에서 메토트렉세이트치료를 병용 투여 받거나 병용 투여 받지 않은 환자에서도 반응이 비슷하였다.
최소 3% 체표면적(body surface area; BSA)이 건선과 관련된 환자에서 건선 면적 및 중증도 지수(Psoriatic Area and Severity Index; PASI) 반응을 평가하였다. 24주차에 PASI가 75% 또는 90% 개선에 도달한 환자의 비율은 이 약 투여군(N=69)에서 각각 59%와 42%였고, 이에 비해 위약군(N=69)에서는 각각 1%와 0%였다(p0.001). PASI 반응은 첫 방문 시점(2주차)에 일부 환자에서 명백하였다. 베이스라인에서 메토트렉세이트 치료를 병용 투여 받거나 병용 투여 받지 않은 환자에서도 반응이 비슷하였다.
표 9: 위약대조 건선성 관절염 시험에서의 ACR 반응(환자 비율)
|
반응 |
위약 N=162 |
이 약 N=151 |
위약 N=49 |
이 약 N=51 |
|
|
PsA 시험 I |
PsA 시험 II | ||
|
ACR 20 |
|
|
|
|
|
12주차 |
14% |
58%a |
16% |
39%b |
|
24주차 |
15% |
57%a |
N/A |
N/A |
|
ACR 50 |
|
|
|
|
|
12주차 |
4% |
36%a |
2% |
25%a |
|
24주차 |
6% |
39%a |
N/A |
N/A |
|
ACR 70 |
|
|
|
|
|
12주차 |
1% |
20%a |
0% |
14%b |
|
24주차 |
1% |
23%a |
N/A |
N/A |
a 이 약과 위약 사이의 모든 비교에 대해 p0.001
b 이 약과 위약 사이의 모든 비교에 대해 p0.05
N/A 해당 없음
표 10: 건선성 관절염 PsA 시험 I에서의 질병 활성도 구성요소
|
매개변수: 중앙값 |
베이스라인 |
24주차 |
베이스라인 |
24주차 |
|
|
위약 N=162 |
이 약* N=151 | ||
|
통증이 있는 관절 수a |
23.0 |
17.0 |
20.0 |
5.0 |
|
종창 관절 수b |
11.0 |
9.0 |
11.0 |
3.0 |
|
의사의 전반적 평가c |
53.0 |
49.0 |
55.0 |
16.0 |
|
환자의 전반적 평가c |
49.5 |
49.0 |
48.0 |
20.0 |
|
통증c |
49.0 |
49.0 |
54.0 |
20.0 |
|
장애 척도(HAQ)d |
1.0 |
0.9 |
1.0 |
0.4 |
|
CRPe |
7.8 |
7.4 |
8.0 |
2.1 |
* 중앙값 변화를 기초로 이 약 vs. 위약 비교에 대해 p0.001
a 0 ‑78 척도
b 0 ‑76 척도
c 시각적 아날로그 척도. 0점 = 최고, 100점 = 최악
d 건강 평가 설문조사의 장애 척도. 0점 = 최고, 3점 = 최악. 옷 입기/치장하기, 일어나기, 먹기, 걷기, 뻗기, 잡기, 위생 유지 및 일상 활동 유지 등을 수행할 수 있는 환자의 능력 측정
e 정상 범위: 0 ‑2.87mg/L
최대 136주 동안의 공개 연장시험에서 ACR 반응은 유지되었다.
건선성 관절염 시험에서 방사선학적 변화를 평가하였다. 손, 손목 및 발의 방사선 사진은 환자가 이 약 또는 위약을 투여 받은 이중눈가림 기간의 베이스라인과 24주차, 그리고 모든 환자가 공개 연장 시험의 이 약을 투여 받은 48주차에 수집하였다. 원위지절간관절을 포함한 수정된 전체 샤프 점수(modified Total Sharp Score; mTss)(즉, 류마티스 관절염에서 사용한 TSS와 동일하지 않음)를 사용하였다.
이 약 치료는 위약 치료와 비교하여 말초 관절 손상의 진행률을 감소시켰고, 이는 베이스라인으로부터 mTss 점수(평균 ± SD) 변화로 측정하였으며 위약군의 경우 0.8 ± 2.5 (24주차), 이 약 투여군의 경우 0.0 ± 1.9 (48주차)였다(p0.001).
베이스라인으로부터 48주차까지 방사선학적 진행이 없는 이 약 투여군(n=102) 중, 84%는 144주간의 치료 동안 방사선학적 진행을 보이지 않았다.
삶의 질 및 신체적 기능
이 약을 투여 받은 환자는 24주차에 위약과 비교하여 HAQ 및 약식 건강 조사(SF ‑36) 평가 결과 신체적 기능에 있어서 통계적으로 유의한 개선이 확인되었다. 개선된 신체적 기능은 최대 136주차의 공개 연장 시험동안 지속되었다.
축성 척추관절염 임상시험
강직성 척추염
활동성 강직성 척추염(ankylosing spondylitis; AS)이 있는 성인 환자 393명을 대상으로 2건의 무작위배정, 24주 이중눈가림, 위약대조 시험에서 이 약 40 mg의 격주 투여에 대한 안전성과 유효성을 평가하였다. 2건 중 규모가 큰 시험(AS 시험 I 또는 M03 ‑607)에는 활동성 강직성 척추염이 있고 기존의 치료에 부적절한 반응을 보인 성인 환자 315명이 등록되었다. 활동성 강직성 척추염은 다음의 3가지 기준 중 최소 2가지를 충족하는 것으로 정의한다: (1) 강직성 척추염 질병 활성도 지수(Bath AS disease activity index; BASDAI] 점수 ≧4 cm, (2) 전체 요통에 대한 시각적 아날로그 점수(visual analog score; VAS) ≧40 mm, (3) 조조 강직 ≧1시간. 눈가림 기간 후 공개 연장시험 기간 동안 환자는 최대 236주의 추가 기간 동안 이 약 40mg을 격주로 피하투여 받았다.
그 결과 이 약을 투여 받은 환자는 위약을 투여 받은 환자와 비교하여 강직성 척추염의 징후 및 증상이 통계적으로 유의하게 개선되었다. 그림 4와 표 11 및 12에 제시한 바와 같이, 질병 활성도 측정으로 확인한 유의한 개선은 2주차에 처음 관찰되었고 24주까지 유지되었다.
전체 척추 강직증이 있는 환자는 이 시험에 포함되었다(n=11). 이 환자들의 반응은 전체 강직증이 없는 환자들과 유사하였다.
표 11: 위약대조 AS 시험에서의 유효성 결과 ‑ AS 시험 I 징후 및 증상 감소
|
반응 |
위약 N=107 |
이 약 N=208 |
|
ASASa 20 |
|
|
|
2주차 |
16% |
42%*** |
|
12주차 |
21% |
58%*** |
|
24주차 |
19% |
51%*** |
|
ASAS 50 |
|
|
|
2주차 |
3% |
16%*** |
|
12주차 |
10% |
38%*** |
|
24주차 |
11% |
35%*** |
|
ASAS 70 |
|
|
|
2주차 |
0% |
7%** |
|
12주차 |
5% |
23%*** |
|
24주차 |
8% |
24%*** |
|
BSADAIb 50 |
|
|
|
2주차 |
4% |
20%*** |
|
12주차 |
16% |
45%*** |
|
24주차 |
15% |
42%*** |
***,** 2, 12, 24주차에서 이 약과 위약 사이의 모든 비교에 대해 p0.001, p0.01에서 통계적으로 유의함
a 강직성 척추염에서의 평가
b 강직성 척추염 활동 지수
위약을 투여 받은 환자 6%와 비교하여 이 약을 투여 받은 환자 22%가 24주차에 낮은 수준의 질병 활성도(4개의 ASAS 반응 매개변수 각각의 수치가 20 [0 ‑100mm 척도])에 도달하였다 (p0.001).
표 12: 강직성 척추염 질병 활성도의 구성요소
|
|
베이스라인 평균 |
24주차 평균 |
베이스라인 평균 |
24주차 평균 |
|
|
위약 N=107 |
이 약 N=208 | ||
|
ASAS 20 반응 기준* |
|
|
|
|
|
환자의 전반적 질병 활성도 평가a* |
65 |
60 |
63 |
38 |
|
전체 요통* |
67 |
58 |
65 |
37 |
|
염증b* |
6.7 |
5.6 |
6.7 |
3.6 |
|
BASFIc* |
56 |
51 |
52 |
34 |
|
BASDAId 점수* |
6.3 |
5.5 |
6.3 |
3.7 |
|
BASMIe 점수* |
4.2 |
4.1 |
3.8 |
3.3 |
|
이주부터 벽까지의 거리(cm) |
15.9 |
15.8 |
15.8 |
15.4 |
|
요추 굴곡(cm) |
4.1 |
4.0 |
4.2 |
4.4 |
|
목 회전(각도) |
42.2 |
42.1 |
48.4 |
51.6 |
|
요추방향 굴곡(cm) |
8.9 |
9.0 |
9.7 |
11.7 |
|
복사뼈 사이의 거리(cm) |
92.9 |
94.0 |
93.5 |
100.8 |
|
CRPf* |
2.2 |
2.0 |
1.8 |
0.6 |
a 시각적 아날로그 척도(VAS) 0점 = “없음” 및 100점 = “중증”으로 측정 시에, 최소 20% 및 10점이 개선된 대상자의 비율
b BASDAI(‘d’에서 정의) 5번 및 6번 질문의 평균
c 강직성 척추염 기능 지수
d 강직성 척추염 질병 활성도 지수
e 강직성 척추염 운동능력 지수
f C ‑반응성 단백질(mg/dL)
* 24주차에 이 약과 위약 사이의 모든 비교에 대해 통계적으로 유의함
2건의 시험 중 규모가 작은 무작위, 이중눈가림, 위약대조 시험(AS 시험 II, M03 ‑606)의 강직성 척추염 환자 82명에서도 비슷한 경향이 확인되었다.
환자 보고 결과는 2건의 강직성 척추염 시험 모두에서 일반적인 건강 상태 설문조사 SF ‑36 및 질병 특이적 강직성 척추염 삶의 질 설문조사(Ankylosing Spondylitis Quality of Life Questionnaire; ASQoL)를 사용하여 평가하였다. 12주차의 SF ‑36 신체적 구성요소 점수는 이 약을 투여 받은 환자(평균 변화 6.93)가 위약을 투여 받은 환자(평균 변화 1.55, p0.001)에 비해 유의하게 개선되었으며 이는 24주차까지 유지되었다(평균 변화 7.44 vs. 1.85).
ASQoL의 결과는 전반적인 삶의 질 개선을 입증하는 이 결과를 뒷받침한다. 이 약을 투여 받은 환자(평균 변화 ‑3.15, p0.001)는 위약을 투여 받은 환자(평균 변화 ‑0.95, p0.001)와 비교하여 12주차에 통계적으로 유의하게 개선되었으며, 이는 24주차까지 유지되었다(평균 변화 ‑3.58 vs ‑1.06).
비방사선학적 축성 척추관절염(방사선학적으로 강직성 척추염이 확인되지 않는 축성 척추관절염)
비방사선학적 축성 척추관절염(non ‑radiographic axial spondyloarthritis; nr ‑asSpA) 환자를 대상으로 2건의 무작위, 이중눈가림, 위약대조 시험에서 이 약의 안전성 및 유효성을 평가하였다. nr ‑axSpA I 시험에서는 활동성 nr ‑axSpA가 있는 환자를 평가하였다. nr ‑axSpA II 시험은 공개 연장 이 약 치료 동안 질병 관해에 도달한 활동성 nr ‑axSpA 환자를 대상으로 한 치료 중단 시험이었다.
nr ‑axSpA I 시험
무작위배정, 12주 이중눈가림, 위약대조 시험인 nr ‑axSpA I 시험에서는 활동성 nr ‑axSpA(질병 활성도의 평균 베이스라인 점수[강직성 척추염 질병 활성도 지수(BASDAI)]가 이 약 투여군의 경우 6.4, 위약군의 경우 6.5) 환자 185명을 대상으로 이 약 40 mg 격주 투여를 평가하였으며, 이 환자들은 ≧1개의 NSAID에 대해 부적절한 반응 또는 불내성이 있거나 NSAID가 금지된 환자였다. 여기에 포함된 환자들은 ASAS 축성 SpA 기준에 따라 분류되었고, 강직성 척추염에 대해 수정된 뉴욕 기준을 충족하는 환자 및 건선이나 건선성 관절염이 있는 환자는 제외하였다. 일차 유효성 평가변수는 12주차에 ASAS 40 반응에 도달한 환자 비율이었다.
베이스라인에서 33명(18%)의 환자는 질병 조절 항류마티스제를 병용 투여 받았고 146명(79%)은 NSAID를 병용 투여 받았다. 이중눈가림 기간 후 공개 연장 기간 동안 환자는 최대 144주간 추가적으로 이 약 40 mg을 격주 피하투여 받았다. 12주차 결과에 따르면 전체 치료군 및 양성 MRI 또는 증가된 CRP를 보인 환자 모두에서 위약과 비교하여 이 약을 투여 받은 환자의 활동성 nr ‑axSpA 징후 및 증상이 통계적으로 유의하게 개선되었다(표 13). nr ‑axSpA의 징후 및 증상이 감소했음을 입증한 변수는 24주차 및 68주차에서도 지속되거나 계속 개선되었으며 156주차까지 유지되었다.
표 13: 위약대조 시험 nr ‑axSpA I에서의 유효성 결과#
|
이중눈가림 12주차에서의 반응 |
위약 N=94 |
이 약 N=91 |
|
ASASa 40 |
15% |
36%*** |
|
ASAS 20 |
31% |
52%** |
|
ASAS 5/6 |
6% |
31%*** |
|
ASAS 부분 관해 |
5% |
16%* |
|
BASDAIb 50 |
15% |
35%** |
|
ASDASc,d,e |
-0.3 |
-1.0*** |
|
ASDAS 비활성 질병 |
4% |
24%*** |
|
SF-36 PCSd,f |
2.0m |
5.5** |
|
HAQ-Sd,g |
-0.1 |
-0.3* |
|
hs-CRPd,h,i |
-0.3 |
-4.7*** |
|
SPARCCj MRI 천장 관절d,k |
-0.6 |
-3.2** |
|
SPARCC MRI 척추d,l |
-0.2 |
-1.8** |
|
a 국제 척추관절염 학회의 평가 b 강직성 척추염 질병 활성도 지수 c 강직성 척추염 질병 활성도 점수 d 베이스라인으로부터의 평균 변화 e 위약 n=91, 이 약 n=87 f 약식-36 건강 상태 설문조사TM 버전 2 신체적 구성요소 요약 점수 g 척추관절증에 대해 수정한 건강 평가 설문조사 h 고감도 C-반응 단백질(mg/L) i 위약 n=73, 이 약 n=70 j 캐나다의 척추관절염 연구 컨소시엄 k 위약 및 이 약 n=84 l 위약 n=82, 이 약 n=85 m n=93 *** p값 < 0.001 ** p값 < 0.01 * p값 < 0.05 # HAQ-S와 hs-CRP의 경우 이전 관찰치 적용(Last observation carried forward; LOCF) 분석, SF-36과 SPARCC MRI 점수의 경우 관찰된 사례 분석, 기타 모든 범주형 변수의 경우 비응답자 결측값 대체(non-responder imputation; NRI) 분석 | ||
염증의 억제
이 약을 투여 받은 환자에서 hs ‑CRP와, 천장관절 및 척추의 MRI로 측정한 염증 징후의 유의한 개선은 각각 156주차와 104주차까지 유지되었다. 천장관절에 대한 SPARCC MRI는 131명의 환자에서 이용 가능했고 척추에 대한 SPARCC MRI는 130명의 환자에서 이용 가능했으며 베이스라인으로부터 104주차까지의 평균 변화는 각각 ‑3.8 및 ‑1.4였다.
삶의 질 및 신체적 기능
건강 관련 삶의 질 및 신체적 기능은 HAQ ‑S 및 SF ‑36 설문조사를 사용하여 평가하였다. 이 약은 위약과 비교하여 베이스라인으로부터 12주차까지 HAQ ‑S 전체 점수 및 SF ‑36 신체적 구성요소 요약(Physical Component Summary; PCS)에 대해 통계적으로 유의하게 큰 개선을 보였다. SF ‑36 PCS 점수 및 HAQ ‑S 전체 점수 결과는 각각 52주차, 68주차 및 156주차까지 지속되었다.
nr ‑axSpA II 시험
≧2개의 NSAID에 대해 부적절한 반응을 보이거나 NSAID에 불내성 또는 금지 사유가 있는 활동성 nr ‑axSpA(평균 베이스라인 질병 활성도[BASDAI]가 7.0) 환자 673명은 nr ‑axSpA II 공개 연장 시험의 표지 기간에 등록되어 28주간 격주로 이 약 40mg을 투여 받았다. 이 환자들은 천장관절이나 척추의 염증에 대해 MRI 또는 증가된 hs ‑CRP와 같은 객관적 증거를 가지고 있었다. 공개 연장 시험 기간 동안 최소 12주차에 지속 관해에 도달한 환자(N=305)(16, 20, 24 및 28주차 ASDAS 1.3)는 이중눈가림, 위약대조 기간(총 시험 기간 68주)의 추가 40주 동안 이 약 40mg 격주 투여군(N=152) 또는 위약군(N=153)으로 무작위 배정되었다. 이중눈가림 기간 동안 flared대상자는 최소 12주 동안의 이 약 40mg 격주 구조요법이 허용되었다.
일차 유효성 평가변수는 시험의 68주차까지 재발이 없는 환자 비율이었다. 재발은 4주 간격의 2회 연속 방문에서 ASDAS ≧ 2.1로 정의하였다. 이 약을 투여 받은 대다수의 환자는 위약을 투여 받은 환자와 비교하여 이중눈가림 기간 동안 질병 재발이 없었다(70.4% vs. 47.1%, p0.001)(그림 5).
그림 5: nr ‑axSpA II에서 재발까지의 시간을 요약한 카플란 ‑메이어 곡선
치료 중단에 배정된 그룹에서 재발한 환자 68명 중 65명은 12주간의 이 약 구조요법을 완료하였고 이 중 37명(56.9%)은 공개 연장 시험을 재개하고 12주 후에 다시 질병 관해에 도달하였다(ASDAS 1.3).
68주까지 지속적으로 이 약을 투여 받은 환자는 이중눈가림 기간 동안 치료 중단으로 배정된 환자와 비교하여 활동성 nr ‑axSpA의 징후 및 증상에 대해 통계적으로 유의하게 큰 개선을 보였다(표 14).
표 14: nr ‑axSpA II 위약대조 기간에서의 유효성 결과
|
이중눈가림 68주차에서의 반응 |
위약 N=153 |
이 약 N=152 |
|
ASASa,b 20 |
47.1% |
70.4%*** |
|
ASASa,b 40 |
45.8% |
65.8%*** |
|
ASASa 부분 관해 |
26.8% |
42.1%** |
|
ASDASc 비활성 질병 |
33.3% |
57.2%*** |
|
부분 재발d |
64.1% |
40.8%*** |
|
a 국제 척추관절염 학회의 평가 b 환자가 활동성 질병을 가지고 있는 경우 베이스라인은 공개라벨 베이스라인으로 정의한다. c 강직성 척추염 질병 활성도 점수 d 부분 재발은 연속 2회 방문에서 ASDAS가 ≥1.3이고 <2.1인 것으로 정의한다. *** p값 < 0.001 ** p값 < 0.01 | ||
크론병
무작위배정, 이중눈가림, 위약대조 시험에서 중등증부터 중증의 활동성 크론병(크론병 활성도 지수[Crohn’s Disease Activity Index; CDAI]가 ≧220 및 ≦450)이 있는 성인 환자를 대상으로 이 약 다중 투여의 안전성과 유효성을 평가하였다. 안정적 용량의 아미노살리실산염, 코르티코스테로이드 및/또는 면역조절제의 투여가 허용되었으며 79%의 환자는 이러한 약물 중 최소 하나의 투여를 지속하였다.
2건의 시험에서 임상적 관해(CDAI 150의 정의)의 유도를 평가하였다. CD 시험 I (M02 ‑403)에서 TNF 차단제 투여 이력이 없는 환자 299명은 다음의 치료군 중 하나로 무작위 배정되었다: 0주차와 2주차에 위약을 투여 받는 위약군, 0주차에 이 약 160 mg과 2주차에 이 약 80 mg을 투여 받는 160/80 투여군, 0주차에 이 약 80 mg과 2주차에 이 약 40 mg을 투여 받는 80/40 투여군, 0주차에 이 약 40mg과 2주차에 이 약20 mg을 투여 받는 40/20 투여군. 임상 결과는 4주차에 평가하였다.
두 번째 유도 시험인 CD 시험 II (M04 ‑691)에서, 인플릭시맙 치료에 반응을 보이지 않거나 불내성이 있는 325명의 환자는 0주차에 이 약 160 mg과 2주차에 이 약 80 mg을 투여 받거나 0주차와 2주차에 위약을 투여 받는 집단으로 무작위 배정되었다. 임상 결과는 4주차에 평가하였다.
CD 시험 III (M02 ‑404)에서 임상적 관해의 유지를 평가하였다. 이 시험에서 활동성 질병이 있는 환자 854명은 공개라벨 이 약을 0주차에 80 mg, 2주차에 40 mg 투여 받았다. 그런 다음 환자는 4주차에 격주 40 mg 이 약 투여군, 매주 40 mg 이 약 투여군, 또는 위약군으로 무작위 배정되었다. 전체 시험 기간은 56주였다. 4주차에 임상 반응을 보인 환자(CR ‑70 = CDAI ≧70 감소)는 4주차에 임상 반응을 보이지 않은 환자와 별도로 계층화하여 분석하였다.
임상적 관해의 유도
TNF 차단제 투여 이력이 없거나(CD 시험 I), 또는 인플릭시맙 치료에 반응을 보이지 않거나 불내성이 있는 것(CD 시험 II)과는 상관 없이 160/80 mg 이 약을 투여 받은 환자의 대부분은 4주차에 위약과 비교하여 임상적 관해를 유도하였다(표 15 참조).
표 15: CD 시험 I 및 CD 시험 II에서 임상적 관해의 유도(환자 비율)
|
|
CD 시험 I |
CD 시험 II | ||
|
|
위약 N=74 |
이 약 160/80mg N=76 |
위약 N=166 |
이 약 160/80mg N=159 |
|
4주차 |
|
|
|
|
|
임상적 관해 |
12% |
36%* |
7% |
21%* |
|
임상 반응 |
34% |
58%** |
34% |
52%** |
|
임상적 관해는 CDAI 점수 <150으로 정의한다. 임상적 반응은 최소 70점의 CDAI 감소로 정의한다. * 비율에 대한 이 약 vs. 위약 쌍비교, p <0.001 ** 비율에 대한 이 약 vs. 위약 쌍비교, p <0.01 | ||||
임상적 관해의 유지
CD 시험 III의 4주차에 환자의 58%(499명/854명)가 임상 반응을 보였고 일차 분석에서 평가되었다. 4주차에서 임상 반응을 보인 환자의 대부분이 26주차와 56주차의 위약 유지군 환자와 비교하여 이 약 40 mg 격주 유지군에서 임상적 관해에 도달하였다(표 16 참조). 매주 이 약 치료를 받은 집단은 이 약을 격주로 투여 받은 집단과 비교하여 유의하게 높은 관해율을 입증하지 못했다.
표 16: CD 시험 III에서 임상적 관해의 유지(환자 비율)
|
평가변수 |
위약 N=170 |
격주 40mg 이 약 N=172 |
매주 40mg 이 약 N=157 |
|
26주차 | |||
|
임상적 관해 |
17% |
40%* |
47%* |
|
임상 반응 |
28% |
54%* |
56%* |
|
56주차 | |||
|
임상적 관해 |
12% |
36%* |
41%* |
|
임상 반응 |
18% |
43%* |
49%* |
임상적 관해는 CDAI 점수 150으로 정의한다. 임상적 반응은 최소 70점의 CDAI 감소로 정의한다.
* 비율에 대한 이 약 vs. 위약 쌍비교, p0.001
시험 동안 관해에 도달한 4주차 반응을 보인 환자 중 격주로 이 약을 투여 받은 환자가 위약 유지군의 환자보다 더 장기간 관해를 유지하였다. 56주차에 질병과 관련된 입원 및 수술은 위약과 비교하여 이 약을 투여 받은 환자에서 통계적으로 유의하게 감소하였다.
CD 시험 I의 환자 117명/276명과 CD 시험 II 및 III의 환자 272명/777명은 최소 3년의 공개라벨 이 약 치료 동안 추적되었다. 각각 88명(75.2%)과 189명(69.5%)의 환자는 임상적 관해가 지속되었다. 임상 반응(CR ‑70)은 각각 107명(91.5%)과 248명(91.2%)의 환자에서 유지되었다.
117명/854명(CD 시험 III)은 스크리닝과 베이스라인 시점 모두에서 배출 누공(draining fistula)을 가지고 있었다. 누공 치유의 평가를 위해 시험에서 사용한 아달리무맙의 두 투여량 모두에 대한 데이터를 통합하였다. 26주차에 누공 치유된 환자의 비율은 아달리무맙을 투여 받은 환자(21명/70명[30.0%])가 위약을 투여 받은 환자(6명/47명[12.8%])에 비해 통계적으로 유의하게 높았다. 완전한 누공 치유는 아달리무맙 투여군과 위약 투여군의 각각 23명/70명(32.9%)과 6명/47명(12.8%)에서 56주차까지 유지되었다.
135명의 환자가 등록된 내시경 시험(M05 ‑769)에서 점막 치유에 대한 이 약의 영향을 확인하였다. 12주차의 점막 치유는 이 약 투여군 중 27.4%, 위약 투여군 중 13.1%에서 확인되었고(p=0.056) 52주차의 점막 치유는 이 약을 투여 받은 환자 중 24.2%, 위약을 투여 받은 환자 중 0%에서 확인되었다(p0.001).
삶의 질
CD 시험 I과 CD 시험 II에서 이 약 80/40 mg 및 160/80 mg 투여군으로 무작위 배정된 환자는 위약군 환자에 비해 질병 특이적 염증성 장질환 설문조사(inflammatory bowel disease questionnaire; IBDQ) 전체 점수에서 통계적으로 유의한 개선을 보였으며, CD 시험 III의 경우 26주차와 56주차에 위약군 환자와 비교하여 아달리무맙 치료군 사이에서도 이러한 개선이 관찰되었다.
궤양성 대장염
2건의 무작위, 이중눈가림, 위약대조 시험에서 중등증부터 중증의 활동성 궤양성 대장염(Mayo 점수 6 ‑12점, 내시경 항목 점수 2 ‑3점)이 있는 성인 환자를 대상으로 이 약 다중 투여의 안전성과 유효성을 평가하였다. 안정적 용량의 아미노살리실산염, 코르티코스테로이드 및/또는 면역조절제의 병용 투여가 허용되었다.
UC ‑I 시험에서 임상적 관해의 유도(Mayo ≦2점, >1점인 항목별 점수는 없는 것으로 정의)를 평가하였다. UC ‑I 시험에서 TNF 길항제 투여 이력이 없는 환자 390명은 0주차와 2주차에 위약 투여군, 0주차에 160 mg 이 약투여 후 2주차에 80 mg 투여군, 또는 0주차에 80 mg 이 약 투여 후 2주차에 40 mg 투여군으로 무작위 배정되었다. 2주차 후에 두 아달리무맙 투여군의 환자는 아달리무맙을 격주로 40 mg 투여 받았다. 임상적 관해는 8주차에 평가되었다.
UC ‑II 시험에서 248명의 환자는 0주차에 160 mg 이 약, 2주차에 80 mg 및 이후 격주로 40 mg 투여 받았으며 246명의 환자는 위약을 투여 받았다. 임상 결과는 8주차에 관해의 유도, 52주차에 관해의 유지에 대해 평가하였다.
160/80 mg 이 약을 투여 받은 대상자는 위약과 비교하여 UC ‑I 시험(각각 18% vs. 9%, p=0.031)과 UC ‑II 시험(각각 17% vs. 9%, p=0.019)에서 통계적으로 유의하게 큰 비율로 8주차에 임상적 관해에 도달하였다. UC ‑II 시험의 8주차에 관해에 도달했던 이 약 투여군 중 21명/41명(51%)은 52주차에 관해를 유지하였다.
전체 UC ‑II 시험군과 전체 Mayo 점수당 치료 8주차에 반응한 환자의 결과는 표 17에 제시한다.
표 17: UC ‑II 시험에서 반응, 관해 및 점막 치유(환자 비율)
|
|
위약
|
격주 40 mg 이 약
|
이 약 160/80/40 mg 8주차 반응자 |
|
|
N=246 |
N=248 |
N=125 |
|
52주차 | |||
|
임상 반응 |
18% |
30%* |
47% |
|
임상적 관해 |
9% |
17%* |
29% |
|
점막 치유 |
15% |
25%* |
41% |
|
≥90일 동안 스테로이드 사용 없는 관해율a |
6% (N=140) |
13%* (N=150) |
20% (N=90) |
|
8주차 및 52주차 | |||
|
지속적 반응 |
12% |
24%** |
- |
|
지속적 관해 |
4% |
8%* |
- |
|
지속적 점막 치유 |
11% |
19%* |
- |
|
임상적 관해는 Mayo 점수가 ≤2이고 >1인 항목별 점수가 없는 것으로 정의한다. * 비율에 대한 이 약 vs. 위약 쌍비교, p <0.05 ** 비율에 대한 이 약 vs. 위약 쌍비교, p <0.001 a 베이스라인에서 코르티코스테로이드를 투여 받은 환자 중 | |||
UC I와 UC II 시험의 통합 분석에서 입원의 모든 원인 및 UC와 관련된 입원율이 통계적으로 유의하게 감소했음을 관찰하였다.
UC ‑II 시험에서 약 40%의 환자가 인플릭시맙을 통한 항 ‑TNF 치료에 실패한 이력이 있었다. 이러한 환자에서 아달리무맙의 효능은 항 ‑TNF 투여 이력이 없는 환자와 비교하여 감소하였다. 항 ‑TNF 치료에 실패 이력이 있는 환자 중 52주차 관해율은 위약군에서 3%, 아달리무맙군에서 10%에 도달하였다.
UC 시험 I 및 II의 환자는 공개라벨 장기간 확장 시험(UC ‑III)으로 전환할 수 있는 선택권이 있었다. 3년간의 이 약 치료 후 74%(268명/360명)의 환자에서 부분 Mayo 점수당 임상적 관해가 지속되었고 최소 4년간 이 약 치료를 받은 환자의 경우 75%(97명/130명)가 부분 Mayo 점수당 임상적 관해가 지속되었다. 1년 이상의 치료 후 반응 도달에 실패한 환자는 매주 40 mg으로 투여 빈도를 증가시킴으로써 치료 이익을 받을 수 있었다.
삶의 질
UC 시험 II에서 이 약 160/80 mg 투여군으로 무작위 배정된 환자는 위약과 비교하여 52주차에 질병 특이적 염증성 장질환 설문조사(IBDQ) 전체 점수의 개선에 도달하였다(p=0.007).
베체트 장염
기존의 치료(스테로이드 또는 면역억제제)에 부적절한 반응을 보인 장내 베체트병이 있는 일본인 환자 대상의 공개, 비통제(uncontrolled) 시험에서 24주차에 현저한 개선율은 45.0%(9명/20명)였다. 현저한 개선은, 종합적인 질병 평가 척도를 사용하여 위장 증상의 전반적 평가와 내시경 평가 점수 모두 ≦1점인 환자의 비율로 정의하였고 이는 궤양의 크기 및 위장 증상 개선을 둘 다 반영한다.
판상 건선
무작위배정, 이중눈가림 시험에서 전신 치료 또는 광선치료의 대상자인 만성 판상 건선( ≧10% BSA 관련, 건선 영역 및 중증도 지수[Psoriasis Area and Severity Index; PASI] ≧12 또는 ≧10)이 있는 성인 환자에서 이 약의 안전성과 유효성을 시험하였다. 건선 시험 I과 II에 등록된 환자 중 73%는 전신 치료나 광선치료를 받은 이력이 있었다. 또한 무작위 배정된 이중눈가림 시험(건선 시험 III)에서 전신 치료의 대상자인 손 및/또는 발의 건선을 동반한 중등증부터 중증의 만성 판상형 건선이 있는 성인 환자에서도 이 약의 안전성과 유효성을 시험하였다.
건선 시험 I (M03 ‑656)은 3개의 치료 기간 동안 1,212명의 환자를 평가하였다. A 기간에 환자는 위약을 투여 받거나 초기 용량 80 mg의 이 약을 투여 받은 후 일주일 뒤부터 격주로 40 mg을 투여 받았다. 16주간의 치료 후 최소 PASI 75 반응(베이스라인과 비교하여 최소 75%의 PASI 점수 개선)에 도달한 환자는 B 기간에 진입하여 공개라벨 40 mg 이 약을 격주로 투여 받았다. 33주차에 ≧PASI 75 반응이 유지되고 기존에 A 기간에서 활성 치료로 무작위 배정된 환자는 C 기간에 무작위 재배정되어 추가 19주 동안 격주로 40 mg 이 약 또는 위약을 투여 받았다. 모든 치료군에서 평균 베이스라인 PASI 점수는 18.9점이었고 베이스라인에서 의사의 전반적 평가(Physician’s Global Assessment; PGA) 점수의 범위는 “중등증”(환자의 53%) ‑“중증”(41%) ‑“매우 중증”(6%)이었다.
건선 시험 II (M04 ‑716)에서는 271명의 환자에서 메토트렉세이트 및 위약에 대해 이 약의 안전성과 유효성을 비교하였다. 환자는 위약, 메토트렉세이트 초기 용량인 7.5mg을 투여 받은 후 12주차까지 최대 25 mg으로 증량하여 투여 받거나, 이 약 초기 용량 80 mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 40 mg을 투여 받았다. 16주 이상의 치료를 통해 이 약과 메토트렉세이트를 비교한 이용가능한 데이터는 없다. MTX를 투여 받고 8주차 및/또는 12주차에 ≧PASI 50 반응에 도달한 환자는 추가로 투여량을 증량하지 않았다. 모든 치료군에서 평균 베이스라인 PASI 점수는 19.7점이었고 베이스라인 PGA 점수의 범위는 “경증”(1%) ‑“중등증”(48%) ‑“중증”(46%) ‑“매우 중증”(6%)이었다.
임상2상 및 임상3상 건선 시험에 모두 참여한 환자는 이 약을 추가로 최소 108주 동안 투여 받는 공개라벨 확장 시험(M03 ‑658)에 등록할 수 있었다.
건선 시험 I과 II에서 일차 평가변수는 베이스라인으로부터 16주차에 PASI 75 반응에 도달한 환자의 비율이었다(표 18 및 19 참조).
표 18: 16주차의 Ps 시험 I (REVEAL) 유효성 결과
|
|
위약 N=398 n(%) |
격주 이 약 40mg N=814 n(%) |
|
≥PASI 75a |
26 (6.5) |
578 (70.9)b |
|
PASI 100 |
3 (0.8) |
163 (20.0)b |
|
PGA: 개선/최소 |
17 (4.3) |
506 (62.2)b |
|
a PASI 75 반응에 도달한 환자 비율은 중심이 보정된 비율로 계산하였다. b p <0.001, 이 약 vs. 위약 | ||
표 19: 16주차의 Ps 시험 II (CHAMPION) 유효성 결과
|
|
위약 N=53 n(%) |
MTX N=110 n(%) |
격주 이 약 40mg N=108 n(%) |
|
≥PASI 75 |
10 (18.9) |
39 (35.5) |
86 (79.6)a,b |
|
PASI 100 |
1 (1.9) |
8 (7.3) |
18 (16.7)c,d |
|
PGA: 개선/최소 |
6 (11.3) |
33 (30.0) |
79 (73.1)a,b |
|
a p <0.001, 이 약 vs. 위약 b p <0.001, 이 약 vs. 메토트렉세이트 c p <0.01, 이 약 vs. 위약 d p <0.05, 이 약 vs. 메토트렉세이트 | |||
건선 시험 I에서 PASI 75 반응에 도달하여 33주차에 위약으로 무작위 재배정된 환자의 28%는 이 약을 지속적으로 투여 받은 환자의 5%와 비교하여(p0.001) “적절한 반응의 실패(loss)”을 경험하였다(베이스라인과 비교하여 PASI 50 반응을 나타내고 33주차와 비교하여 PASI 점수가 6점 이상 감소한, 33주 후 52주차 이하의 PASI 점수). 위약으로 무작위 재배정된 후 적절한 반응 도달에 실패하여 공개라벨 확장 시험에 등록된 환자 중 38% (25명/66명)과 55% (36명/66명)는 각각 12주와 24주의 재치료를 받은 후 PASI 75 반응에 다시 도달하였다.
16주차와 33주차의 PASI 75 반응자 총 233명은 건선 시험 I에서 52주 동안 지속적으로 이 약을 투여 받았으며 공개라벨 확장 시험에서 이 약을 지속적으로 투여 받았다. 이 환자들에서 PASI 75와 PGA 개선 또는 최소 반응률은 공개라벨에서의 추가 108주 치료 후 각각 74.7%와 59.0%였다(총 160주).
건선 시험 II에서 이 약 치료로 무작위 배정된 총 94명은 공개라벨 확장 시험에서 이 약을 지속적으로 투여 받았다. 이 환자들에서 PASI 75와 PGA 개선 또는 최소 반응률은 공개라벨에서의 추가 108주 치료 후 각각 58.1%와 46.2%였다(총 124주).
총 347명의 안정적인 반응자가 공개라벨 확장 시험에서 중단 및 재치료 평가에 참여하였다. 재발까지의 시간 중앙값(PGA가 “중등증” 또는 최악으로 감소)은 약 5개월이었다. 중단 기간 동안 반동현상(rebound)을 경험한 환자는 없었다. 재치료 기간에 등록된 환자 중 76.5% (218명/285명)은 중단 동안의 재발 여부와 관계 없이 재치료 16주 후 PGA “개선” 또는 “최소” 반응을 보였다(중단 기간 동안 재발 또는 재발되지 않은 환자에서 각각 69.1% [123명/178명], 88.8% [95명/107명]).
위약(시험 I 및 II) 및 MTX(시험 II)와 비교하여 베이스라인으로부터 16주차에 DLQI(피부학적 삶의 질 지수)에서 유의한 개선이 입증되었다. 시험 I에서 SF ‑36의 신체적 및 정신적 구성요소 요약 점수 또한 위약과 비교하여 유의하게 개선되었다.
공개라벨 확장 시험에서 PASI 반응이 50% 미만으로 감소하여 격주 40mg에서 매주 40mg으로 투여량을 증량한 환자 중 각각 26.4% (92명/349명)와 37.8% (132명/349명)는 12주차와 24주차에 PASI 75 반응에 도달하였다.
건선 시험 III (REACH)은 중등증부터 중증의 만성 판상형 건선과 함께 손 및/또는 발 건선이 있는 72명의 환자를 대상으로 위약에 대해 이 약의 효능 및 안전성을 비교하였다. 환자는 이 약 초기 용량 80mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 이 약 40mg을 투여 받거나 위약을 투여 받았다. 16주차에 통계적으로 유의하게 많은 비율의 이 약 투여군 환자가 위약군 환자에 비해 손 및/또는 발에 대해 PGA ‘개선’ 또는 ‘거의 개선’에 도달하였다(각각 30.6% vs. 4.3% [P=0.014]).
건선 시험 IV는 중등증부터 중증의 손발톱 건선이 있는 성인 환자 217명에서 위약에 대해 이 약의 안전성과 유효성을 비교하였다. 환자는 이 약 초기 용량 80mg을 투여 받은 후 일주일 뒤부터 16주 동안 격주로 이 약 40mg을 투여 받거나 위약을 투여 받았고, 추가 26주 동안 공개라벨 이 약 치료를 받았다. 손발톱 건선은 수정된 손발톱 건선 중증도 지수(mNAPSI) 및 손톱 건선에 대한 의사의 전반적 평가(PGA ‑F)를 사용하여 평가하였다. 위약에 무작위 배정된 환자와 비교하여 이 약에 무작위 배정된 환자 중 통계적으로 유의하게 높은 비율의 환자가 26주차에 mNAPSI에서 최소 75% 개선(mNAPSI 75)에 도달하였다(표 20 참조). NAPSI의 개선율은 이 약 투여군 환자가 위약군 환자보다 16주차(44.2% vs.7.8%)와 26주차(56.2% vs 11.5%)에 통계적으로 유의하게 높았다.
이 약 투여군에서 통계적으로 유의하게 높은 비율의 환자는 위약군과 비교하여 26주차에 베이스라인으로부터 2등급 이상 개선하여 PGA ‑F “개선” 또는 “최소”에 도달하였다. 이 시험에서 이 약은 다양한 범위(BSA ≧10%와 BSA 10% 및 ≧5%)의 손발톱 건선 환자에서 치료 이익을 입증하였으며 위약과 비교하여 두피 건선에서도 통계적으로 유의한 개선을 보였다.
표 20: 26주차의 유효성 결과
|
평가변수 |
위약 N=108 |
격주 이 약 40mg N=109 |
|
≥mNAPSI 75(%) |
3.4 |
46.6a |
|
PGA-F 개선/최소 및 2등급 이상 개선(%) |
6.9 |
48.9a |
|
전체 손발톱 NAPSI 변화율(%) |
-11.5 |
-56.2a |
|
mNAPSI = 0 (%) |
0 |
6.6b |
|
손발톱 통증 수치 등급 척도의 변화 |
-1.1 |
-3.7a |
|
손발톱 건선 신체적 기능 중증도 점수의 변화 |
-0.8 |
-3.7a |
|
B-SNIPI 50 두피(%) |
N=12 0.4 |
N=18 58.3b |
|
a p <0.001, 이 약 vs. 위약 b p <0.05, 이 약 vs. 위약 B-SNIPI 50: 베이스라인 두피 점수보다 6점 이상 증가한 대상자 중 Brigham 두피 손톱 역위 손-발바닥 건선 지수(Brigham Scalp Nail Inverse Palmo-Plantar Psoriasis index; B-SNIPI)의 두피 구성요소가 최소 50% 감소 | ||
52주차까지 이 약 치료를 지속적으로 투여 받은 환자 중 65.0%가 mNAPSI 75 반응에 도달하였고 61.3%는 PGA ‑F 반응에 도달하였다.
이 약을 투여 받은 환자는 위약과 비교하여 26주차의 DLQI(피부학적 삶의 질 지수)가 베이스라인으로부터 통계적으로 유의하게 개선되었다. 26주차에 베이스라인으로부터의 평균 감소(개선)는 이 약군(N=94)에서 8.0, 위약군(N=93)에서 1.9였다.
화농성 한선염
전신 항생제 치료에 불내성 또는 부적절한 반응을 보이거나 금지 사유가 있는 중등증에서 중증의 화농성 한선염(hidradenitis suppurativa; HS) 성인 환자를 대상으로 무작위, 이중눈가림, 위약대조 시험에서 이 약의 안전성과 유효성을 평가하였다. HS ‑I 시험과 HS ‑II 시험의 환자는 최소 3개의 농양 또는 염증성 결절이 있는 Hurley II단계 또는 III단계 질병을 가지고 있었다.
HS ‑I 시험(M11 ‑313)은 2개의 치료 기간에서 307명의 환자를 평가하였다. A 기간에서 환자는 0주차에 위약 또는 이 약 초기 용량 160 mg, 2주차에 80 mg, 4주차부터 11주차까지 매주 40mg을 투여 받았다. 항생제 병용 투여는 시험 동안 허용되지 않았다. 12주간의 치료 후 A 기간에 이 약을 투여 받은 환자는 B 기간에 3개의 치료군 중 하나로 무작위 재배정되었다(12주차부터 35주차까지 매주 이 약 40mg 투여군, 격주 이 약 40mg 투여군, 또는 위약군). A 기간에 위약으로 무작위 배정된 환자는 B 기간에 매주 이 약 40mg 투여군으로 배정하였다.
HS ‑II 시험(M11 ‑810)은 2개의 치료 기간에서 326명의 환자를 평가하였다. A 기간에서 환자는 0주차에 위약 또는 이 약 초기 용량 160 mg, 2주차에 80 mg, 4주차부터 11주차까지 매주 40 mg을 투여 받았다. 환자 중 19.3%는 시험 동안 베이스라인의 경구 항생제 치료를 지속하였다. 12주간의 치료 후 A 기간에 이 약을 투여 받은 환자는 B 기간에 3개의 치료군 중 하나로 무작위 재배정되었다(12주차부터 35주차까지 매주 이 약 40 mg 투여군, 격주 이 약 40 mg 투여군, 또는 위약군). A 기간에 위약으로 무작위 배정된 환자는 B 기간에 위약군으로 배정하였다.
HS ‑I 시험과 HS ‑II 시험에 참여한 환자는 이 약 40 mg을 매주 투여 받는 공개라벨 확장 시험에 등록될 수 있었다. 모든 아달리무맙 투여군의 평균 노출은 762일이었다. 3개의 치료군 모두 국소 소독 세척을 매일 실행하였다.
임상 반응
염증 병변의 임상 반응은 화농성 한선염 임상 반응(HiSCR: 전체 농양 및 염증성 결절 수가 최소 50% 감소하고 베이스라인과 비교하여 농양 수가 증가하거나 배출 누공의 수가 증가하지 않음)을 사용하여 평가하였다. HS와 관련된 피부 통증의 감소는 11점 척도에서 초기 베이스라인 점수가 3점 이상이었던 환자에서 수치 등급 척도를 사용하여 평가하였다.
12주차에 위약과 비교하여 유의하게 높은 비율의 이 약 투여 환자가 HiSCR에 도달하였다. 12주차에 HS ‑II 시험에서 유의하게 높은 비율의 환자는 HS와 관련된 피부 통증이 임상적으로 의미 있게 감소됨을 경험하였다(표 21 참조). 이 약을 투여 받은 환자는 치료 초기 12주 동안 질병 재발의 위험이 유의하게 감소하였다.
표 21: HS 시험 I과 II의 12주차에서의 유효성 결과
|
|
HS 시험 I |
HS 시험 II | ||
|
평가변수 |
위약 |
매주 이 약 40mg |
위약 |
매주 이 약 40mg |
|
화농성 한선염 임상 반응(HiSCR)a |
N=154 40 (26.0%) |
N=153 64 (41.8%)* |
N=163 45 (27.6%) |
N=163 96 (58.9%)*** |
|
피부 통증 ≥30% 감소b |
N=109 27 (24.8%) |
N=122 34 (27.9%) |
N=111 23 (20.7%) |
N=105 48 (45.7%)*** |
|
* p <0.05, 이 약 vs. 위약 *** p <0.001, 이 약 vs. 위약 a 무작위 배정된 모든 환자 b 수치 등급 척도 0-10점을 기초로 베이스라인에서 HS와 관련된 피부 통증 평가 ≥3점인 환자. 0점 = 피부 통증 없음, 10점 = 상상할 수 있는 최악의 피부 통증 | ||||
지속적으로 이 약을 매주 투여 받는 집단에 무작위 배정된 환자의 12주차 전체 HiSCR 비율은 96주차까지 유지되었다. 96주 동안 매주 이 약 40 mg을 장기간 투여 받은 환자에서 새로운 안전성 결과는 확인되지 않았다.
위약과 비교하여 베이스라인으로부터 12주차의 큰 개선은 피부학적 삶의 질 지수(DLQI)로 측정한 피부 특이적 건강 관련 삶의 질(HS ‑I 시험 및 HS ‑II 시험), 치료 만족 설문조사 ‑ 약물(TSQM)로 측정한 환자의 전반적인 약물 치료 만족(HS ‑I 시험 및 HS ‑II 시험), 그리고 SF ‑36의 신체적 구성요소 요약 점수로 측정한 신체적 건강(HS ‑I 시험)으로 입증하였다.
포도막염
독립적인 앞포도막염 환자를 제외하고 비감염성 중간포도막염, 후포도막염 및 전체포도막염(“후부에 영향을 미치는 비감염성 포도막염”으로도 알려짐)이 있는 성인 환자를 대상으로 2건의 무작위, 이중눈가림, 위약대조 시험(UV 시험 I [M10 ‑877] 및 UV 시험 II [M10 ‑880])에서 이 약의 안전성과 유효성을 평가하였다. 환자는 위약을 투여 받거나 초기 용량 80mg의 이 약을 투여 받은 후 일주일 뒤부터 격주로 40mg을 투여 받았다. 안정적 용량의 비생물학적 면역억제제를 병용 투여하는 것은 허용되었다. 두 시험의 일차 효능 평가변수는 ‘치료 실패까지의 시간’이었다. 초기 질병 제어 이후 치료 실패까지의 시간을 연장하면 질병 재발, 염증 및 시력 상실의 위험이 감소된다.
치료 실패는 염증성 맥락망막 및/또는 염증성 망막 혈관 병변, 전방(anterior chamber; AC) 세포 등급, 유리체 혼탁(vitreous haze; VH) 등급 및 최대교정시력(best corrected visual acuity; BCVA)를 기반으로 복합적인 구성요소 결과를 통해 정의하였다.
UV 시험 I은 코르티코스테로이드(10 ‑60mg/일의 경구 프레드니손) 치료에도 불구하고 활동성 포도막염이 있는 환자 217명을 평가하였다. 모든 환자는 시험 등록시 프레드니손의 표준 용량 60mg/일을 투여 받은 후 의무적으로 테이퍼링(tapered) 계획에 따라 코르티코스테로이드를 15주차까지 중단하였다.
UV 시험 II는 베이스라인에서 질병 제어를 위해 만성 코르티코스테로이드(10 ‑35 mg/일의 경구 프레드니손) 치료가 필요한 비활동성 포도막염 환자 226명을 평가하였다. 환자는 이후 의무적으로 테이퍼링 계획에 따라 코르티코스테로이드를 19주차까지 중단하였다.
임상 반응
두 시험 결과는 이 약을 투여 받은 환자가 위약을 투여 받은 환자와 비교하여 치료 실패의 위험이 통계적으로 유의하게 감소했음을 입증하였다(표 22 참조). 두 시험은 위약과 비교하여 치료 실패율에 대한 이 약의 초기에 지속적인 영향을 입증하였다(그림 6 참조).
표 22: UV 시험 I과 II의 치료 실패까지의 시간
|
분석 치료 |
N |
실패 N(%) |
치료 실패까지의 시간(개월) |
HRa |
HRa에 대한 95% CI |
P값b |
|
UV 시험 I에서 6주차 또는 그 이후 치료 실패까지의 시간 | ||||||
|
일차 분석(ITT) | ||||||
|
위약 |
107 |
84 (78.5) |
3.0 |
- |
- |
- |
|
아달리무맙 |
110 |
60 (54.5) |
5.6 |
0.50 |
0.36, 0.70 |
<0.001 |
|
UV 시험 II에서 2주차 또는 그 이후 치료 실패까지의 시간 | ||||||
|
일차 분석(ITT) | ||||||
|
위약 |
111 |
61 (55.0) |
8.3 |
- |
- |
- |
|
아달리무맙 |
115 |
45 (39.1) |
NEc |
0.57 |
0.39, 0.84 |
0.004 |
|
참고: 6주차 이후의 치료 실패(UV 시험 I) 또는 2주차 이후의 치료 실패(UV 시험 II)는 사건으로 계산되었다. 치료 실패 외의 이유로 인한 중단은 중단 시점에서 제외되었다. a 치료를 인자로 설정한 비례 위험 회귀 모델의 아달리무맙 vs. 위약 HR b 로그 순위 검정의 양측 P값 c NE = 추정할 수 없음. 위험 환자의 절반 미만이 사건을 경험하였다. | ||||||
그림 6: 6주차 이후(UV 시험 I) 또는 2주차 이후(UV 시험 II)
치료 실패까지의 시간을 요약한 카플란 ‑메이어 곡선
두 시험 모두에서 일차 평가변수의 모든 요소는 이 약군과 위약군 사이의 전반적인 차이에 점증적으로 기여하였다(표 23).
표 23: UV 시험 I과 II의 치료 실패 구성 요소
|
|
UV I |
UV II | ||||
|
치료 실패까지의 시간의 구성 요소 |
HRa |
95% CI |
P값b |
HRa |
95% CI |
P값b |
|
새로운 활동성 염증성 병변 |
0.38 |
(0.21-0.69) |
0.001 |
0.55 |
(0.26-1.15) |
0.105 |
|
전방 세포 등급 |
0.51 |
(0.30-0.86) |
0.01 |
0.7 |
(0.42-1.18) |
0.18 |
|
유리체 혼탁 등급 |
0.32 |
(0.18-0.58) |
<0.001 |
0.79 |
(0.34-1.81) |
0.569 |
|
최대교정시력 |
0.56 |
(0.32-0.98) |
0.04 |
0.33 |
(0.16-0.70) |
0.002 |
|
참고: 6주차 이후의 치료 실패(UV 시험 I) 또는 2주차 이후의 치료 실패(UV 시험 II)는 사건으로 계산되었다. 치료 실패 외의 이유로 인한 중단은 중단 시점에서 제외되었다. a 치료를 인자로 설정한 비례 위험 회귀 모델의 아달리무맙 vs. 위약 HR b 로그 순위 검정의 양측 P값 | ||||||
또한 UV 시험 I에서 AC 세포 등급, 유리체 혼탁 등급 및 logMAR BCVA(6주차 전의 최적 상태에서 마지막 방문까지의 평균 변화, P값은 각각 0.011, 0.001 및 0.003)의 변화에 대해 위약과 비교하여 아달리무맙에 유리하게 통계적으로 유의한 차이가 관찰되었다.
UV 시험 I 및 UV 시험 II의 비통제 장기간 확장에 포함된 417명의 환자 중 46명이 부적합한 것으로 간주되었고(예: 당뇨망막병증에 의한 합병증 진행, 백내장 수술 또는 유리체절제술로 인해) 효능의 일차 분석에서 제외되었다. 나머지 371명의 환자 중 276명의 평가 가능한 환자는 78주간의 공개라벨 아달리무맙 치료를 완료하였다. 관찰된 데이터 접근법을 기반으로, 222명(80.4%)은 매일 ≦7.5mg의 스테로이드 투여를 동반한 무활동기였고(활동성 염증성 병변 없음, AC 세포 등급 ≦0.5+, VH 등급 ≦0.5+) 184명(66.7%)은 스테로이드를 투여하지 않는 무활동기였다. BCVA는 78주차에 눈의 88.4%에서 개선 또는 유지되었다(5문자 미만으로 하락). 78주 전에 시험을 중단한 환자 중 11%는 이상사례, 5%는 아달리무맙 치료에 대한 불충분한 반응으로 인해 중단하였다.
삶의 질
UV 시험 I에서 이 약 치료는 NEI VFQ ‑25로 측정한 시력과 관련된 기능 및 건강 관련 삶의 질을 유지시켰다.
⓶ 소아
소아 특발성 관절염
다관절성 소아 특발성 관절염(pJIA)
다양한 소아 특발성 관절염 발생 유형(가장 흔한 류마티스인자 음성 또는 양성 다관절성 및 확장성 소수 관절염)을 가진 활동성 다관절성 또는 다관절 과정 소아 특발성 관절염이 있는 소아를 대상으로 2건의 시험(pJIA I 및 II)에서 이 약의 안전성과 유효성을 평가하였다.
pJIA I
다관절성 소아 특발성 관절염(juvenile idiopathic arthritis; JIA)이 있는 171명의 아동(4 ‑ 17세)을 대상으로 다기관, 무작위, 이중눈가림, 평행군 시험에서 이 약의 안전성과 유효성을 평가하였다. 공개라벨 유도 단계(open ‑label lead in; OL LI)에서 환자는 메토트렉세이트를 투여 받은 환자 또는 메토트렉세이트를 투여 받지 않은 환자의 두 집단으로 계층화되었다. 비 ‑메토트렉세이트층에 있는 환자는 메토트렉세이트 투여 이력이 없거나 시험 약물을 투여하기 최소 2주 전에 MTX를 중단한 환자였다. 환자는 안정적 용량의 NSAID 및/또는 프레드니손( ≦0.2 mg/kg/일 또는 최대 10 mg/일)의 투여를 유지하였다. OL LI 단계에서 모든 환자는 24 mg/m2, 최대 40 mg의 이 약을 16주 동안 격주로 투여 받았다. 연령별 환자 분포 및 OL LI 단계 동안 투여 받은 최소, 중앙값, 최대 용량은 표 24에 제시한다.
표 24: 연령별 환자 분포와 OL LI 단계 동안 투여 받은 이 약 용량
|
연령군 |
베이스라인 환자 수 n(%) |
최소, 중앙값 및 최대 용량 |
|
4 - 7세 |
31 (18.1) |
10, 20 및 25mg |
|
8 - 12세 |
71 (41.5) |
20, 25 및 40mg |
|
13 - 17세 |
69 (70.4) |
25, 40 및 40mg |
16주차에 소아 ACR 30 반응을 입증한 환자는 이중눈가림(double blind; OB) 단계에서 무작위 배정되어 추가 32주 동안, 또는 질병이 재발할 때까지 이 약 24 mg/m2, 최대 40 mg을 투여 받거나 위약을 격주로 투여 받았다. 질병 재발 기준은 6개의 소아 ACR 핵심 기준 중 3개 이상이 베이스라인으로부터 ≧30% 악화되고, ≧2개의 활동성 관절 및 6개의 기준 중 >30%의 개선을 보이는 기준이 1개를 넘지 않는 것으로 정의하였다. 32주 후 또는 질병 재발 시 환자는 공개라벨 연장 단계에 등록될 수 있었다.
표 25: JIA 시험에서 소아 ACR 30 반응
|
계층 |
MTX |
비-MTX | ||
|
단계 |
|
| ||
|
OL-LI 16주 |
|
| ||
|
소아 ACR 30 반응(n/N) |
94.1% (80/85) |
74.4% (64/86) | ||
|
| ||||
|
이중눈가림 |
이 약 (n=38) |
위약 (n=37) |
이 약 (n=30) |
위약 (n=28) |
|
32주 마지막에서의 질병 재발a (n/N) |
36.8% (14/38) |
64.9% (24/37)b |
43.3% (13/30) |
71.4% (20/28)c |
|
질병 재발까지의 시간 중앙값 |
>32주 |
20주 |
>32주 |
14주 |
|
a 48주차 소아 ACR 30/50/70 반응은 위약을 투여 받은 환자보다 유의하게 높았다. b p=0.015 c p=0.031 | ||||
16주차에 반응을 보인 환자(n=144) 중, 시험 전반에 걸쳐 이 약을 투여 받은 환자는 OLE 단계에서 최대 6년간 ACR 30/50/90 반응이 유지되었다. 전체적으로 19명의 대상자는 6년 이상 이 약을 투여 받았으며 이들 중 11명은 베이스라인 연령군 4 ‑ 12세, 8명은 베이스라인 연령군 13 ‑ 17세였다.
이 약과 메토트렉세이트를 병용 투여했을 경우 이 약을 단독 투여했을 때보다 전반적인 반응은 일반적으로 우수했으며 보다 적게 항체가 발생하였다. 이러한 결과를 고려해볼 때, 이 약을 메토트렉세이트와 병용 투여하는 것이 권장되며 메토트렉세이트의 투여가 적절하지 않은 환자는 단일 요법으로 사용하는 것이 권장된다.
pJIA II
중등증부터 중증의 활동성 다관절성 JIA가 있는 32명의 아동(2세 ‑4세 아동 또는 체중이 15kg인 4세 이상의 아동)을 대상으로 공개라벨, 다기관 시험에서 이 약의 안전성과 유효성을 평가하였다. 환자는 24 mg/m2 체표면적(body surface area; BSA), 최대 20 mg의 이 약을 최소 24주 동안 SC 단일 주입을 통해 격주로 투여 받았다. 시험 동안 대부분의 대상자는 MTX를 병용 투여하고 있었으며 일부에서 코르티코스테로이드 또는 NSAID 병용 투여를 보고하였다.
12주차와 24주차에 관찰된 데이터 접근법을 사용한 소아 ACR 30 반응은 각각 93.5%와 90.0%였다. 12주차와 24주차에 소아 ACR 50/70/90에 도달한 대상자의 비율은 각각 90.3%/61.3%/38.7%와 83.3%/73.3%/36.7%였다. 24주차에 반응(소아 ACR 30)을 보인 환자(30명 중 27명) 중 이 기간 동안 이 약을 투여 받은 환자는 OLE 단계에서 최대 60주 동안 ACR 30 반응이 유지되었다. 전반적으로 20명의 대상자가 60주 이상 이 약을 투여 받았다.
골부착부염 관련 관절염
골부착부염 관련 관절염이 있는 46명의 소아 환자(6 ‑ 17세)를 대상으로 다기관, 무작위, 이중눈가림 시험에서 이 약의 안전성과 유효성을 평가하였다. 환자는 24 mg/m2 체표면적(body surface area; BSA), 최대 40 mg의 이 약 또는 위약을 12주 동안 투여하는 집단 중 하나로 무작위 배정되었다. 이중눈가림 기간 후 공개라벨(open ‑label; OL) 기간 동안 환자는 이 약 24 mg/m2 BSA, 최대 40 mg을 추가 192주 동안 격주로 피하 투여 받았다. 일차 평가변수는 베이스라인부터 12주차까지 관절염(기형 또는 통증 및/또는 압통을 동반한 운동 상실 관절로 인한 것이 아닌 부종)이 있는 활동성 관절 수의 변화율이었으며 이 약군의 평균 비율 감소는 ‑62.6%인 반면 위약군에서는 ‑11.6%였다. 관절염이 있는 활동성 관절 수의 개선은 공개라벨 기간 동안 156주차까지 유지되었다. 환자의 대다수는 부착부염 부위의 수, 압통 관절 수(tender joint count; TJC), 종창 관절 수(swollen joint count; SJC), 소아 ACR 30 반응, 소아 ACR 50 반응 및 소아 ACR 70 반응과 같은 이차 평가변수의 임상적 개선을 입증하였으며, 이러한 개선은 공개라벨 기간 동안 156주차까지 유지되었다.
소아 크론병
소아 크론병 활동도 지수(Pediatric Crohn's Disease Activity Index; PCDAI) 점수가 >30인 것으로 정의되는 중등증부터 중증의 크론병(Crohn’s disease; CD)이 있는 6 ‑ 17세의 소아 환자 192명을 대상으로 체중(40 kg 또는 ≧40 kg)에 따른 이 약의 유도 및 유지 치료에 대한 효능 및 안전성을 평가하기 위해 다기관, 무작위, 이중눈가림 임상시험을 설계하여 이 약을 평가하였다. 대상자는 CD에 대한 기존 치료(코르티코스테로이드 및/또는 면역조절제 포함)에 실패한 이력이 있어야 했다. 또한 대상자는 인플릭시맙에 대한 반응 도달에 실패했거나 불내성이 있을 수 있다.
모든 대상자는 베이스라인 체중을 기반으로 한 투여량으로 공개라벨 유도 치료를 받았다. 체중이 ≧40 kg인 환자의 경우 0주차에 160 mg, 2주차에 80 mg을 투여 받았으며 40 kg인 환자의 경우에는 각각 80 mg과 40 mg을 투여 받았다.
4주차에 대상자는 표 26에 제시된 바와 같이 저용량 또는 표준 용량 유지 요법을 받는 시점의 체중에 따라 각 투여군에 1:1로 무작위 배정되었다.
표 26: 유지 요법
|
환자 체중 |
저용량 |
표준용량 |
|
<40kg |
격주 10mg |
격주 20mg |
|
≥40kg |
격주 20mg |
격주 40mg |
유효성 결과
시험의 일차 평가변수는 26주차에 PCDAI 점수 ≦10점으로 정의되는 임상적 관해였다.
임상적 관해 및 임상 반응(PCDAI 점수가 베이스라인으로부터 최소 15점 감소되는 것으로 정의) 비율은 표 27에 제시한다. 코르티코스테로이드 또는 면역조절제의 중단율은 표 28에 제시한다.
표 27: 소아 CD 시험의 PCDAI 임상적 관해 및 반응
|
|
표준용량 격주 40/20mg N=93 |
저용량 격주 20/10mg N=95 |
P값* |
|
26주차 |
|
|
|
|
임상적 관해 |
38.7% |
28.4% |
0.075 |
|
임상 반응 |
59.1% |
48.4% |
0.073 |
|
52주차 |
|
|
|
|
임상적 관해 |
33.3% |
23.2% |
0.100 |
|
임상 반응 |
41.9% |
28.4% |
0.038 |
|
* 표준용량 vs 저용량 비교에 대한 p값 | |||
표 28: 소아 CD 시험의 코르티코스테로이드 또는 면역조절제 중단 및 누공 관해
|
|
표준용량 격주 40/20mg |
저용량 격주 20/10mg |
P값1 |
|
코르티코스테로이드 중단 |
N=33 |
N=38 |
|
|
26주차 |
84.8% |
65.8% |
0.066 |
|
52주차 |
69.7% |
60.5% |
0.420 |
|
면역조절제 중단2 |
N=60 |
N=57 |
|
|
52주차 |
30.0% |
29.8% |
0.983 |
|
누공 관해3 |
N=15 |
N=21 |
|
|
26주차 |
46.7% |
38.1% |
0.608 |
|
52주차 |
40.0% |
23.8% |
0.303 |
|
1 표준용량 vs 저용량 비교에 대한 p값 2 면역조절제 치료는 대상자가 임상 반응 기준을 충족했을 때 시험자의 재량으로 26주차 이후에만 중단할 수 있었다. 3 최소 2번의 연속적인 베이스라인 후 방문 동안, 베이스라인에서 배출이 진행되었던 모든 누공의 폐쇄로 정의 | |||
26주차와 52주차에 두 치료군에서 체질량지수 및 신장 속도에 대해 베이스라인으로부터 통계학적으로 유의한 증가(개선)가 관찰되었다. 삶의 질 매개변수(IMPACT III 포함)에 대해서도 두 치료군 모두에서 베이스라인으로부터 통계적 및 임상적으로 유의한 개선이 관찰되었다.
소아 CD 시험의 환자는 공개라벨 장기간 확장 시험에서 투여를 지속할 수 있는 선택권이 있었다. 5년간의 이 약 치료 이후 환자의 74% (37명/50명)은 임상적 관해가 지속되었고 환자의 92% (46명/50명)는 PCDAI에 따라 임상 반응이 지속되었다.
소아 판상 건선
국소 치료 및 일광요법이나 광선요법으로 부적절하게 제어된 중증의 만성 판상형 건선(PGA ≧4 또는 >20% BSA 관련 또는 매우 두꺼운 병변을 동반한 >10 % BSA 관련 또는 PASI ≧20 또는 임상적으로 관련된 얼굴, 생식기, 손/발 병변을 동반한 PASI ≧10으로 정의)이 있는 4세 소아 환자 114명을 대상으로 무작위, 이중눈가림, 통제 시험에서 이 약의 효능을 평가하였다.
환자는 격주 0.8 mg/kg(최대 40mg) 이 약, 격주 0.4 mg/kg(최대 20mg) 이 약, 또는 매주 0.1 ‑0.4 mg/kg(최대 25mg) 메토트렉세이트를 투여 받았다. 16주차에 이 약 0.8 mg/kg 투여군에 무작위 배정된 환자가 MTX에 무작위 배정된 환자보다 더 많은 수에서 호의적인 유효성 반응(예: PASI 75)을 보였다.
표 29: 16주차의 소아 판상 건선 유효성 결과
|
|
MTXa N=37 |
이 약 격주 0.8mg/kg N=38 |
|
PASI 75b |
12 (32.4%) |
22 (57.9%) |
|
PGA: 개선/최소c |
15 (40.5%) |
23 (60.5%) |
|
a MTX = 메토트렉세이트 b P=0.027, 이 약 0.8mg/kg vs. MTX c P=0.083, 이 약 0.8mg/kg vs. MTX | ||
PASI 75 및 PGA 개선이나 최소에 도달한 환자는 최대 36주간의 치료를 중단하였으며 질병 제어의 실패(PGA 반응 도달 실패)에 대해 모니터링되었다. 그런 다음 환자는 추가 16주 동안 아달리무맙 0.8 mg/kg을 격주로 재투여하였으며 재투여 동안 관찰된 반응은 이전의 이중눈가림 기간에 관찰된 것과 유사하였다: PASI 75 반응 78.9%(19명 중 15명), PGA 개선 또는 최소 52.6 %(19명 중 10명).
시험의 공개라벨 기간에 PASI 75 및 PGA 개선이나 최소 반응은 새로운 안전성 결과 없이 추가적으로 최대 52주 동안 유지되었다.
⓷ 면역원성
항 ‑아달리무맙 항체의 형성은 아달리무맙의 청소율 증가 및 효능 감소와 관련된다. 항 ‑아달리무맙 항체의 존재와 이상반응 사이의 상관관계는 명백하지 않다.
면역원성 분석이 의약품마다 다르기 때문에 다른 의약품의 항체 비율과 비교하는 것은 오해의 소지가 있을 수 있다.
성인
류마티스 관절염 시험 I, II 및 III의 환자에서 6 ‑12개월 동안 다양한 시점에서 항 ‑아달리무맙 항체에 대해 검사하였다. 확증시험에서 항 ‑아달리무맙 항체가 아달리무맙을 투여 받은 환자의 5.5% (58명/1053명)에서 확인되었고, 그에 반해 위약을 투여 받은 환자에서는 0.5% (2명/370명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 12.4%였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 0.6 %였다.
건선성 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 10 % (38명/376명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 13.5 % (24명/178명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 7 % (14명/198명)였다.
강직성 척추염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 8.3 % (17명/204명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 8.6 % (16명/185명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 5.3 % (1명/19명)였다.
비방사선학적 축성 척추관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 지속적으로 투여 받은 환자의 8명/152명(5.3 %)에서 확인되었다.
크론병 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 2.6% (7명/269명)에서 확인되었다.
중등증에서 중증의 활동성 UC 환자에서 아달리무맙을 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 5.0%였다.
장내 베체트병이 있는 일본인 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 5% (1명/20명)에서 확인되었다.
건선 환자에서 항 ‑아달리무맙 항체는 메토트렉세이트를 병용 투여하지 않은 아달리무맙 투여 환자의 8.4 % (77명/920명)에서 확인되었다. 메토트렉세이트를 병용 투여하지 않고 장기간 아달리무맙을 투여하고 중단 및 재투여 시험에 참여한 판상형 건선 환자에서 재투여 후 항 ‑아달리무맙 항체의 발생률은 2.3 %였고 중단 전에 관찰된 발생률인 1.9%와 유사하였다.
중등증에서 중증의 화농성 한선염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 10.1% (10명/99명)에서 확인되었다.
비감염성 포도막염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 4.8% (12명/249명)에서 확인되었다.
소아
4 ‑ 17세의 다관절성 소아 특발성 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 16%에서 확인되었다. 메토트렉세이트를 병용하지 않은 환자의 경우 발생률은 26%였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 6%였다. 2세부터 4세 또는 체중이 15 kg인 4세 이상의 다관절성 소아 특발성 관절염 환자의 7%에서 항 ‑아달리무맙 항체가 확인되었고 1명의 환자는 메토트렉세이트를 병용 투여하는 중이었다.
골부착부위염 관련 관절염 환자에서 항 ‑아달리무맙 항체는 아달리무맙을 투여 받은 환자의 11% (5명/46명)에서 확인되었다. 메토트렉세이트를 병용 투여 받지 않은 환자의 경우 항체 발생률은 14% (3명/22명)였고 아달리무맙을 메토트렉세이트와 병용 투여한 경우에는 8% (2명/24명)였다.
중등증에서 중증의 활동성 소아 크론병 환자에서 아달리무맙을 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 3.3%였다.
소아 건선 환자에서 0.8 mg/kg 아달리무맙을 단일 투여 받은 환자의 항 ‑아달리무맙 항체 생성률은 13% (5명/38명)였다.
3) 비임상 안전성 자료
비임상 데이터에 따르면 아달리무맙은 단회 투여 독성, 반복 투여 독성 및 유전 독성 시험을 근거로 인간에 대해 특별한 위해성을 나타내지 않는다.
배아 태아 발달 독성/주산기 발달 연구는 사이노몰로거스(cynomologous) 원숭이에서 0, 30 및 100 mg / kg (그룹 당 9 ‑17 마리의 원숭이)의 용량으로 수행되었으며, 아달리무맙이 태아에게 해를 입힌다는 증거는 없었다. 설치류 TNF에 대한 교차 반응이 제한적인 항체 및 설치류에서 중화 항체의 개발에 대한 적절한 모델이 없기 때문에 아달리무맙에 대한 발암성 연구나 생식기능 및 출생 후 독성에 대한 표준 평가는 수행되지 않았다.