
| 제품명 |
|
|||
|---|---|---|---|---|
| 성분 / 함량 |
|
|||
| 첨가제 | ||||
| 도핑금지 약물정보 |
· 경기기간중 : 금지 · 경기기간외 : 허용
상세정보 확인
※ 국소투여경로 등 예외 적용이 있을 수 있으니, KADA 누리집으로 연결되는 "상세정보 확인"을 클릭하여 주시기 바랍니다. |
|||
| 전문 / 일반 |
|
단일 / 복합 | ||
| 제조 / 수입사 | ||||
| 제형 | 투여경로 | |||
| 성상 | ||||
| 허가일 |
|
|||
| 재심사 | ||||
| 대조 / 생동 | ||||
| 급여정보 |
|
|||
| ATC 코드 | ||||
| KPIC 약효분류 |
|
|||
| KPIC 학술 |
팜리뷰
[Social Pharmacy Perspectives] 비만에 관한 사회적 비용과 건강 정책, 약학정보원(대구가톨릭대학교 약학대학 이승미 교수), 2022-02-25
팜리뷰
[Trend Focus] 비만 치료 약제의 최신 동향, 약학정보원(국립암센터 약제부 서다솜), 2022-02-18
팜리뷰
[Drug Safety Report] 비만 치료제의 안전성 정보와 이상사례 사례연구, 약학정보원(대한약사회 지역의약품안전센터), 2022-02-11
팜리뷰
[Pharmacotherapy Today] 비만(obesity) 약물요법, 약학정보원(서울아산병원 약제팀 김수연), 2022-02-04
팜리뷰
음지의 비만(2), 약학정보원(최혁재), 2017-09-11
팜리뷰
음지의 비만(1), 약학정보원(최혁재), 2017-09-04
팜리뷰
비만의 치료 - 약물요법, 약학정보원(송영천), 2014-02-10
팜리뷰
[약물]비만(Obesity)치료의 최신 지견_대한비만학회 2022 진료지침을 중심으로, 약학정보원(김예지 약사), 12 5 2023
|
|||
| 대한약사저널 |
한약제제
비만 <4> 사상의학의 대표적 비만처방 - 흉격이 과열된 환자에 독활지황탕·형방지황탕, 최해륭 약사, 2022-09-26
한약제제
비만 <3> 비만에도 쓸 수 있는 '방풍통성산' - 사람의 표리상하를 시원하게 풀어주는 약, 최해륭 약사, 2022-09-19
한약제제
비만 <2> 빼내야 빠진다, 동의보감 속 해법은? - 잘 쌓이고 잘 나오는 내장지방, 습관이 중요하다, 최해륭 약사, 2022-09-13
한약제제
비만 <1> 운화와 통혈을 주관하는 '비장' - 전통의학에선 비만의 원인을 '비기허'로 보기도, 최해륭 약사, 2022-09-05
이슈트랜드
비만 심할수록 대사증후군 빈도 증가, 2020-08-31
이슈트랜드
비만의 진단과 치료, 2020-08-24
이슈트랜드
비만 약료의 현재와 미래, 약학정보원 학술정보센터, 2020-08-17
약대약
[영양이상 약물] 콘트라브서방정 vs 큐시미아, 정경혜 교수, 2020-01-28
약대약
[비만치료제 약물] 삭센다 vs 큐시미아, 김예지 약사, 2020-01-21
|
|||
| 제품설명서 | 보 기 ( 2017-10-10 게시 ) | |||
| 의약품안전성 정보(DUR) |
||||
| 포장단위 (식약처 기준) |
||||
| 저장방법 | ||||
사용자GNB바
컨텐츠
의약품 상세정보

허가정보 ∙ 복약정보
효능 · 효과
다음의 초기 체질량 지수(BMI)의 성인 환자에서 저칼로리 식이 요법과 만성 체중 관리를 위한 신체 활동 증가의 보조요법으로 사용된다.
◦ 고혈압, 제2형 당뇨병 또는 이상지질혈증과 같은 적어도 하나의 체중 관련 동반질환이 있는 경우 27 kg/m2이상
이 약은 다른 식욕억제제와 병용하지 않고 단독으로만 사용해야 한다.
용법 · 용량
이 약은 음식 섭취와 무관하게 매일 아침에 복용하고, 불면증이 생길 수 있으니 저녁 복용을 피한다.
2) 14일 이후에는 권장량 7.5mg/46mg 용량을 12주간 복용한다.
3) 체중 감량을 확인하여 7.5mg/46mg 복용으로 최초 투여시점 전 체중 대비 3% 이상을 감량하지 못한 경우, 복용을 중단하거나 복용량을 증량한다.
4) 복용량을 증량하는 경우, 11.25mg/69mg 용량을 매일 14일간 복용한다.
5) 14일 이후에는 15mg/92mg 용량을 12주간 복용한다.
6) 체중 감량을 확인하여 15mg/92mg 복용으로 최초 투여시점 전 체중 대비 5% 이상을 감량하지 못한 경우, 복용을 중단한다.
*3.75mg/23mg 과 11.25mg/69mg 용량은 적정을 위해서만 사용한다.
복용 중단
갑작스러운 복용 중단은 발작을 일으킬 가능성이 있기 때문에 치료를 완전히 중단하기 전에 적어도 1주일 동안 하루 걸러 한 번 복용하여 점차적으로 15mg/92mg 용량의 복용을 중단한다.
신장애 환자
중등도 (30 이상 50 mL/min 미만의 크레아티닌 청소율 [CrCl]) 또는 중증 (30 mL/min 미만의 CrCl)의 신장애 환자에서는 1일 1회 7.5mg/46mg을 초과해서는 안 된다.
신장애 환자는 실제 체중과 함께 Cockcroft ‑Gault 계산식을 사용해서 CrCl 계산을 통해 결정된다.
간장애 환자
중등도의 간장애 환자(Child ‑Pugh score 7 ‑9)에서는 1일 1회 7.5mg/46mg을 초과해서는 안 된다.
사용상의 주의사항
1. 경고
2) 태아 독성 : 이 약은 태아에 유해한 영향을 줄 수 있다. 임신 및 역학 관련 연구 자료에 따르면 임신 초기에 이 약의 성분인 토피라메이트에 노출된 태아는 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다.
환자가 임신 중에 이 약을 복용하거나 이 약 복용 중에 임신한 경우, 치료를 즉시 중단하고 태아에게 미칠 잠재적인 위험이 있음을 환자에게 알려야 한다.
가임기 여성은 이 약으로 치료를 시작하기 전에 임신 테스트의 결과가 음성임을 확인해야 하고, 이 약으로 치료하는 동안에는 매달 한 번씩 임신 테스트를 해야 한다. 이를 위해 1회 1개월분 이상의 약을 처방하지 않아야 한다. 가임기 여성은 이 약으로 치료 중에는 효과적인 방법으로 피임을 해야 한다. [임부 및 수유부에 대한 투여 (7.1) 및 (7.4) 참조]
3) 이 약의 성분인 토피라메이트를 투여 받은 환자에서 중대한 피부반응(스티븐스존슨 증후군(SJS) 및 독성표피괴사용해(TEN))이 보고되었다. 대부분의 경우 스티븐슨존슨 증후군(SJS) 및 독성표피괴사용해(TEN)와 연관이 있다고 알려져 있는 다른 약물들을 함께 복용한 환자에서 발생하였으나, 몇몇 사례는 토피라메이트를 단독으로 복용하는 환자에서도 보고되었다. 따라서 중대한 피부반응의 징후를 환자에게 알리는 것이 권고된다. 스티븐슨존슨 증후군(SJS) 또는 독성표피괴사용해(TEN)가 의심될 경우, 이 약의 사용을 중단해야 한다.
2. 다음 환자에는 투여하지 말 것
1) 임신부
2) 녹내장 환자
3) 갑상선 기능 항진증 환자
4) MAO 억제제를 복용중이거나 또는 복용 후 14일이 경과하지 않은 환자 (혈압 상승 위험 유발)
5) 교감신경흥분성 아민에 대한 과민 반응 또는 특이체질 환자
6) 이 약의 성분에 과민증이 있는 환자
7) 18세 미만의 소아
8) 진전된 동맥경화증 환자
9) 심혈관계 질환 환자
10) 중등도 ~ 중증의 고혈압 환자
11) 폐동맥 고혈압 환자
12) 정신적으로 매우 불안하거나 흥분상태에 있는 환자
13) 약물남용의 병력이 있는 환자
3. 다음 환자에는 신중히 투여할 것
1) 신장애 환자
2) 간장애 환자
3) 이 약은 황색4호(타르트라진)를 함유하고 있으므로 이 성분에 과민하거나 알레르기 병력이 있는 환자에는 신중히 투여한다.(큐시미아캡슐7.5mg/46mg, 큐시미아캡슐11.25mg/69mg, 큐시미아캡슐15mg/92mg에 한함)
4) 이 약은 황색5호(선셋옐로우 FCF, Sunset Yellow FCF)를 함유하고 있으므로 이 성분에 과민하거나 알레르기 병력이 있는 환자에는 신중히 투여한다.(큐시미아캡슐7.5mg/46mg, 큐시미아캡슐11.25mg/69mg, 큐시미아캡슐15mg/92mg에 한함)
4. 이상반응
1) 임상시험
임상시험은 광범위하고 다양한 조건에서 시행되므로, 약물의 임상시험에서 관찰되는 이상반응 비율을 다른 약물의 임상시험에서 관찰되는 이상반응 비율과 직접적으로 비교할 수 없으며, 실제로 관찰되는 비율에 반영되지 않을 수도 있다.
이 자료는 평균 298일 동안 노출된 2,318명의 성인 환자(고혈압 환자 936명[40.4%], 제2형 당뇨 환자 309명[13.3%], BMI가 40 kg/m2 초과인 환자 808명[34.9%])를 대상으로 한 2개의 1년, 무작위, 이중 맹검, 위약 대조, 다기관 임상시험과 2개의 2상 임상 보조 연구를 통해 이 약에 대한 노출을 반영하여 기술하였다.
흔한 이상반응 : 5% 이상, 위약에 비해 최소 1.5배 이상으로 나타나는 이상반응으로는 지각 이상, 어지러움, 미각이상, 불면증, 변비, 구강 건조 등이 있다.
이 약의 치료 환자군의 2% 이상 및 위약군에서 보다 빈번하게 보고된 이상 반응은 표 1에 나와 있다.
표 1. 1년의 치료기간 동안 환자의 2% 이상 및 위약군에서 자주 보고된 이상 반응 ‑ 전반적인 연구 개체군 (Overall Study Population)
|
기관계 대분류(System Organ Class, SOC) 대표 용어(Preferred Term, PT) |
위약 (N=1561) % |
이 약 3.75mg/23mg (N=240) % |
이 약 7.5mg/46mg (N=498) % |
이 약 15mg/92mg (N=1580) % |
|
신경계 장애 |
|
|
|
|
|
지각 이상 |
1.9 |
4.2 |
13.7 |
19.9 |
|
두통 |
9.3 |
10.4 |
7.0 |
10.6 |
|
어지러움 |
3.4 |
2.9 |
7.2 |
8.6 |
|
미각이상 |
1.1 |
1.3 |
7.4 |
9.4 |
|
감각저하 |
1.2 |
0.8 |
3.6 |
3.7 |
|
주의력 장애 |
0.6 |
0.4 |
2.0 |
3.5 |
|
정신 장애 |
|
|
|
|
|
불면증 |
4.7 |
5.0 |
5.8 |
9.4 |
|
우울증 |
2.2 |
3.3 |
2.8 |
4.3 |
|
불안 |
1.9 |
2.9 |
1.8 |
4.1 |
|
위장관계 장애 |
|
|
|
|
|
변비 |
6.1 |
7.9 |
15.1 |
16.1 |
|
구강건조 |
2.8 |
6.7 |
13.5 |
19.1 |
|
오심 |
4.4 |
5.8 |
3.6 |
7.2 |
|
설사 |
4.9 |
5.0 |
6.4 |
5.6 |
|
소화불량 |
1.7 |
2.1 |
2.2 |
2.8 |
|
위-식도 역류 질환 |
1.3 |
0.8 |
3.2 |
2.6 |
|
구강 지각 이상 |
0.3 |
0.4 |
0.6 |
2.2 |
|
전신질환 및 투여 부위 상태 |
|
|
|
|
|
피로 |
4.3 |
5.0 |
4.4 |
5.9 |
|
과민성 |
0.7 |
1.7 |
2.6 |
3.7 |
|
갈증 |
0.7 |
2.1 |
1.8 |
2.0 |
|
가슴 불편함 |
0.4 |
2.1 |
0.2 |
0.9 |
|
시각 장애 |
|
|
|
|
|
시야흐림 |
3.5 |
6.3 |
4.0 |
5.4 |
|
안통 |
1.4 |
2.1 |
2.2 |
2.2 |
|
눈 건조 |
0.8 |
0.8 |
1.4 |
2.5 |
|
심장 장애 |
|
|
|
|
|
두근거림 |
0.8 |
0.8 |
2.4 |
1.7 |
|
피부 및 피하조직 장애 |
|
|
|
|
|
발진 |
2.2 |
1.7 |
2.0 |
2.6 |
|
탈모 |
0.7 |
2.1 |
2.6 |
3.7 |
|
신진 대사와 영양 장애 |
|
|
|
|
|
저칼륨혈증 |
0.4 |
0.4 |
1.4 |
2.5 |
|
식욕 감소 |
0.6 |
2.1 |
1.8 |
1.5 |
|
생식 기관 및 유방 질환 |
|
|
|
|
|
월경통 |
0.2 |
2.1 |
0.4 |
0.8 |
|
감염 |
|
|
|
|
|
상기도감염 |
12.8 |
15.8 |
12.2 |
13.5 |
|
비인두염 |
8.0 |
12.5 |
10.6 |
9.4 |
|
부비동염 |
6.3 |
7.5 |
6.8 |
7.8 |
|
기관지염 |
4.2 |
6.7 |
4.4 |
5.4 |
|
인플루엔자 |
4.4 |
7.5 |
4.6 |
4.4 |
|
요로감염 |
3.6 |
3.3 |
5.2 |
5.2 |
|
위장염 |
2.2 |
0.8 |
2.2 |
2.5 |
|
근골격계 및 결합 조직 장애 |
|
|
|
|
|
등통증 |
5.1 |
5.4 |
5.6 |
6.6 |
|
사지 통증 |
2.8 |
2.1 |
3.0 |
3.0 |
|
근육연축 |
2.2 |
2.9 |
2.8 |
2.9 |
|
근육골격통증 |
1.2 |
0.8 |
3.0 |
1.6 |
|
경부통증 |
1.3 |
1.3 |
2.2 |
1.2 |
|
호흡기, 흉부 및 종격동 장애 |
|
|
|
|
|
기침 |
3.5 |
3.3 |
3.8 |
4.8 |
|
굴울혈 |
2.0 |
2.5 |
2.6 |
2.0 |
|
인후두 통증 |
2.0 |
2.5 |
1.2 |
2.3 |
|
비충혈 |
1.4 |
1.7 |
1.2 |
2.0 |
|
상해, 중독 및 시술 후 합병증 |
|
|
|
|
|
시술 후 통증 |
1.7 |
2.1 |
2.4 |
1.9 |
이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 및 위약을 복용한 환자에서 손, 발, 얼굴 따끔거림 등의 지각 이상이 각각 4.2%, 13.7%, 19.9%, 1.9% 발생했다.
미각이상은 금속성 맛(metallic taste)으로 특징되고, 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 투여군에서 각각 1.3%, 7.4%, 9.4% 발생했으며, 이는 위약 투여군에서 1.1% 발생했다. 이러한 증상 대부분은 약물 치료 후 첫 12주 이내에 발생했다. 그러나 일부 환자군에서는 치료하는 동안 나중에 발생하는 것으로 보고되었다. 이 약 치료군에 해당하는 환자의 일부가 이러한 증상으로 인해 치료를 중단했다. (지각 이상의 경우 1%, 미각이상의 경우 0.6%).
기분 장애 및 수면 장애
이 약의 1년간 위약‑대조임상 시험에서 기분 장애 및 수면 장애와 관련된 하나 이상의 이상반응을 보고한 환자의 비율은 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 용량에서 각각 15.8%, 14.5%, 20.6%이었으며, 위약의 경우 10.3%였다. 이 증상들은 수면 장애, 불안 및 우울증으로 분류되었다. 수면 장애에 대한 보고는 일반적으로 불면증의 특징을 보였고, 이 약 3.75mg/23mg, 7.5mg/46mg 및 15mg/92mg 용량으로 치료받은 환자군에서 각각 6.7%, 8.1%, 11.1% 발생했으며, 위약으로 치료받은 환자군의 5.8%에서 발생했다. 불안은 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 용량으로 치료받은 환자군에서 각각 4.6%, 4.8%, 7.9% 발생했으며, 위약으로 치료받은 환자군의 2.6%에서 발생했다. 우울증/기분 장애에 대한 보고는 이 약 3.75mg/23mg, 7.5mg/46mg 및 15mg/92mg 용량으로 치료받은 환자군에서 각각 5.0%, 3.8% 및 7.6% 발생했으며, 위약으로 치료받은 환자군에서 3.4% 발생했다. 이러한 증상 대부분은 약물 치료 후 첫 12주 이내에 발생했다. 그러나 일부 환자군에서는 치료 과정에서 나중에 발생하는 것으로 보고되었다. 이 약의 임상 시험에서 기분과 수면 이상반응의 전반적인 유병률은 우울증 병력이 없는 환자에 비해 우울증의 병력이 있는 환자군에서 약 2배나 높았다. 그러나 기분 및 수면 이상반응이 나타난 환자들의 비율은 이 약으로 치료를 받는 환자군과 위약으로 치료를 받은 환자군에서 비슷하게 나타났다. 우울증 관련 증상은 모든 치료군에서 우울증 과거력이 있는 환자에서 더 빈번하게 발생했다. 그러나 이러한 증상의 발생률에 대한 위약‑보정된 차이(placebo‑adjusted difference)는 과거 우울증 병력에 관계없이 집단 간에 일정하게 유지되었다.
인지 장애
1년간의 위약‑대조임상 시험에서 1개 이상의 인지 관련 이상반응을 겪은 환자의 비율은 3.75mg/23mg 용량에서 2.1%, 7.5mg/46mg 용량에서 5.0%, 15mg/92mg 용량에서 7.6%, 위약에서 1.5% 였다. 이러한 이상반응은 주로 주의/집중력, 기억력, 언어(단어 선택) 문제였다. 이 증상들은 일반적으로 치료 첫 4주 이내에 시작되었고, 평균적으로 약 28일 이하의 기간 동안 지속되었으며, 치료를 중단하면 원상태로 회복되었다. 그러나 특정 환자의 경우 치료 후반 및 장기간동안 이 증상들을 경험했다.
실험실검사수치 이상
⓵ 혈청 중탄산염
1년간의 위약‑대조임상 시험에서 정상 범위 이하(2회 연속 방문 시 또는 최종 방문 시 21 mEq/L 미만의 수준)의 혈청 중탄산염의 지속적인 treatment‑emergent(TE) 감소 발생률은 3.75mg/23mg 투여군에서 8.8%, 7.5mg/46mg 투여군에서 6.4%, 15mg/92mg 투여군에서 12.8%, 위약 투여군에서 2.1%였다. 지속적이고 현저하게 낮은 혈청 중탄산염 수치(2회 연속 방문 시 또는 최종 방문 시 17 mEq/L 미만의 수준) 발생률은 3.75mg/23mg 투여군에서 1.3%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.7%, 위약 투여군에서 0.1% 였다. 일반적으로 혈청 중탄산염 농도의 감소는 경도 수준(평균 1‑3 mEq/L)이었고 치료 초기(4주 방문)에 발생했지만, 치료 후기에 중증 감소(severe decreases) 및 감소(decreases)가 발생하기도 했다.
⓶ 혈청 칼륨
1년간의 위약‑대조임상시험 중 지속적으로 낮은 혈청칼륨 수치(2회 연속 방문 시 또는 최종 방문 시 3.5 mEq/L 미만)의 발생률은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 3.6%, 15mg/92mg 투여군에서 4.9%, 위약 투여군에서 1.1%였다. 지속적으로 낮은 혈청칼륨(low serum potassium)을 경험한 피험자들 중 88%는 비‑칼륨보존이뇨제 (non‑potassium sparing diuretic)로 치료를 받았다.
임상시험기간 동안 현저히 낮은 혈청칼륨(3 mEq/L 미만, 치료 전보다 0.5 mEq/L 이상 감소)의 발생률은 3.75mg/23mg 투여군에서 0.0%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.7%, 위약 투여군에서 0.0%였다. 지속적으로 현저히 낮은 혈청칼륨(3 mEq/L 미만 및 2회 연속 방문시 또는 최종 방문시 치료 전보다 0.5 mEq/L 초과 감소)의 발생률은 3.75mg/23mg 투여군에서 0.0%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.1%, 위약 투여군에서 0.0%였다.
저칼륨혈증은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 1.4%, 15mg/92mg 투여군에서 2.5%, 위약 투여군에서 0.4%였다. 혈중 칼륨 감소는 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 0.4%, 15mg/92mg 투여군에서 1.0%, 위약 투여군에서 0.0%였다.
⓷ 혈청 크레아티닌
1년간의 위약‑대조임상시험에서 기저치 대비 복용량과 관련된 증가가 나타났으며, 4주에서 8주 사이에 정점에 도달했다가 감소하였으나 1년간의 치료 기간 동안 기저치 이상으로 상승 유지되었다. 치료기간 동안 0.3 mg/dL 이상의 혈청 크레아티닌 증가가 발생할 확률은 3.75mg/23mg 투여군에서 2.1%, 7.5mg/46mg 투여군에서 7.2%, 15mg/92mg 투여군에서 8.4%, 위약 투여군에서 2.0%였다. 기저치 대비 50% 이상의 혈청 크레아티닌이 증가한 비율은 3.75mg/23mg 투여군에서 0.8%, 7.5mg/46mg 투여군에서 2.0%, 15mg/92mg 투여군에서 2.8%, 위약 투여군에서 0.6% 였다.
신석증
1년간의 위약‑대조임상시험에서 신장결석증 발생률은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 1.2%, 위약 투여군에서 0.3% 였다.
이상반응에 따른 약물 중단
1년간의 위약‑대조 임상시험에서 3.75mg/23mg 투여군에서 11.6%, 7.5mg/46mg 투여군에서 11.6%, 15mg/92mg 투여군에서 17.4%, 위약 투여군에서 8.4%가 보고된 이상반응으로 인해 약물 치료를 중단했다. 치료중단을 야기한 가장 흔한 이상반응은 표 2에 나타난다.
표 2. 1% 이상에서 약물 치료 중단을 야기한 이상반응(1 year clinical trials)
|
약물치료 중단을 야기한 이상반응a |
위약 (N=1561) % |
이 약 3.75mg/23mg (N=240) % |
이 약 7.5mg/46mg (N=498) % |
이 약 15mg/92mg (N=1580) % |
|
시야흐림 |
0.5 |
2.1 |
0.8 |
0.7 |
|
두통 |
0.6 |
1.7 |
0.2 |
0.8 |
|
과민성 |
0.1 |
0.8 |
0.8 |
1.1 |
|
어지러움 |
0.2 |
0.4 |
1.2 |
0.8 |
|
지각 이상 |
0.0 |
0.4 |
1.0 |
1.1 |
|
불면 |
0.4 |
0.0 |
0.4 |
1.6 |
|
우울증 |
0.2 |
0.0 |
0.8 |
1.3 |
|
불안 |
0.3 |
0.0 |
0.2 |
1.1 |
|
a 치료군 중 어느 하나에서라도1% 이상의 환자가 치료를 중단한 경우 | ||||
미국에서 이 약의 성분인 펜터민과 토피라메이트의 시판후 사용에 대하여 다음과 같은 이상반응이 보고되었다. 이러한 이상반응은 인구수를 특정하기 어려운 모집단으로부터 자발적으로 보고되기 때문에 발생 빈도를 정확하게 예측하거나 약물 노출과의 인과 관계를 확립하는 것이 항상 가능하지는 않다.
큐시미아
정신 장애 : 자살 생각, 자살 행동
안과 질환 : 급성폐쇄각녹내장, 안압 상승
펜터민
알레르기 이상반응 : 두드러기
심혈관 이상반응 : 혈압 상승, 허혈성 사건
중추 신경계 이상반응 : 다행감, 정신증, 진전
생식 이상반응 : 성욕의 변화, 발기부전
토피라메이트
피부질환 : 수포 피부 반응(다형 홍반, 스티븐존슨 증후군, 독성표피괴사용해 포함), 천포창
위장 장애 : 췌장염
간 질환 : 간 실조(사망자 포함), 간염
대사 장애 : 고암모니아혈증, 저체온증
안과 질환 : 황반증
5. 일반적 주의
1) 심박수 증가
이 약은 안정시 심박수를 증가시킬 수 있다.
이 약으로 치료한 과체중 및 비만 성인과 위약으로 치료한 과체중 및 비만 성인을 비교하였을 때, 이 약 치료군에서 기저치에 비해 분당 심박수가 5, 10, 15 및 20회 (bpm) 이상 증가한 환자의 비율이 높았다.
표 3은 최대 1년간의 임상연구에서 심박수의 상승을 보이는 환자의 수와 백분율을 나타낸다.
표 3. 단일 시점을 기준으로 기저치 심박수에서 심박수가 증가한 환자의 수와 백분율
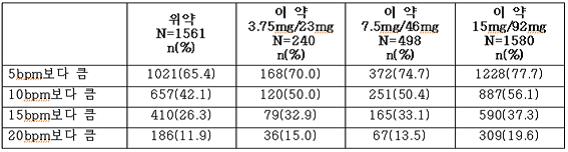
지난 6개월 동안 심근경색이나 뇌졸중 병력이 있는 환자, 생명을 위협하는 부정맥 또는 울혈성심부전증이 있는 환자와 같은 심장 및 뇌혈관 질환 환자에 대해 특히 이 약의 치료로 인한 심박수 상승의 임상적 의미는 분명하지 않다.
이 약을 복용하는 모든 환자, 특히 심장 질환이나 뇌혈관 질환이 있는 환자 또는 이 약의 복용을 시작하거나 늘릴 때 안정시 심박수를 정기적으로 측정할 것을 권장한다. 이 약은 불안정한 심장 질환이나 뇌혈관 질환이 있는 환자를 대상으로 연구를 진행하지 않았으므로 이 환자들에 대한 사용은 권장하지 않는다.
환자는 이 약의 치료기간 동안 휴식을 취할 때 두근거림이나 고동치는 느낌을 경험하는 경우, 이 사실을 의료진에게 알려야 한다. 이 약을 복용하는 동안 안정시 심박수가 지속적으로 증가하는 환자의 경우, 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
2) 자살 행동과 자살 생각
이 약의 성분인 토피라메이트를 포함한 항경련제(AED)는 이러한 약물을 복용하는 목적과 상관없이 약물을 복용하는 환자에게 자살 충동이나 자살 행동 위험을 증가시킨다. 이 약으로 치료받는 환자는 우울증의 발생이나 증상의 악화, 자살 충동이나 자살 행동 및/또는 환자의 기분이나 행동의 비정상적인 변화에 대해 모니터링되어야 하며, 이러한 증상 또는 행동이 발현될 경우 즉시 의료전문가에게 보고될 수 있도록 한다. 자살 충동이나 자살 행동을 경험한 환자는 이 약의 복용을 중단한다.
자살 시도나 적극적인 자살 생각의 병력이 있는 환자는 이 약의 복용을 피해야 한다.
여러 적응증이 있는 11가지 항경련제에 대한 199개의 위약 대조 임상연구(단일요법 및 보조요법, 중간 치료 기간 12주)를 통합 분석한 결과, 항경련제 중 하나에 무작위로 배정된 환자는 자살 충동이나 행동이 위약에 무작위로 배정된 환자에 비해 약 2배의 위험도(보정 상대 위험도(adjusted relative risk) 1.8, 95% 신뢰 구간 [CI] 1.2, 2.7)를 나타냈다. 위약으로 치료한 환자 16,029명에서의 자살 행동이나 자살 생각의 예상 발생률은 0.24%인 것에 비해, 항경련제로 치료한 환자 27,863명에서 예상 발생률은 0.43%로, 치료받은 환자 530명당 약 1건의 자살 생각이나 행동의 증가가 나타났다. 임상시험에서 항경련제로 치료받은 환자에서는 4건의 자살이 있었고 위약으로 치료받은 환자에서는 한 건도 없었지만, 그 수가 너무 적어 자살과 관련된 항경련제 영향에 대해서는 결론을 내릴 수 없었다.
항경련제로 인한 자살 충동이나 행동 위험의 증가는 항경련제로 약물 치료를 시작한 후 빠르면 1주일 후에 관찰되었으며, 치료 기간 내내 지속되었다. 임상분석에 포함된 대부분의 임상시험들은 24주를 초과하지 않았으므로, 24주 이후의 자살 충동이나 행동의 위험을 평가할 수 없었다.
자살 충동이나 행동의 위험은 분석된 데이터에 사용된 약물에서 일반적으로 일치했다. 다양한 작용 기전과 다양한 범위의 적응증을 가진 항경련제에서 위험 증가가 확인되었다는 것은, 항경련제의 적응증과는 무관하게 모든 항경련제에 위험성이 있다는 것을 시사한다. 위험도는 분석된 임상시험에서 연령(5세에서 100세)에 따라 크게 변하지 않았다.
3) 급성 근시 및 2차성 폐쇄각녹내장
이 약의 성분인 토피라메이트로 치료받은 환자들에서 2차성 폐쇄각녹내장과 관련된 급성 근시 증후군이 보고되었으며, 그 증상에는 갑작스런 시력저하 및/또는 안통이 포함된다. 안과적인 검사시 근시, 좁아진 전방, 안 충혈 및 안압 상승이 관찰될 수 있다. 산동은 나타날 수도 있고 아닐 수도 있다. 이 증후군은 모양체상방 삼출 (supraciliary effusion)로 인한 수정체와 홍체의 전방 이동 및 2차성 폐쇄각녹내장과 관련이 있을 수 있다. 증상은 일반적으로 토피라메이트 치료 시작 후 1개월 이내에 발생하지만 치료 중 언제든지 발생할 수 있다. 증상에서 회복되기 위한 1차 치료는 이 약의 복용을 즉각 중단하는 것이다.
어떤 병에 의한 안압상승이라도 치료받지 않으면 영구 시력손실을 포함한 심각한 후유증을 가져올 수 있다.
이 약의 성분인 토피라메이트를 투여 받은 환자에게서 안압 상승과 독립적으로 시야결손이 보고되었다. 임상시험에서 이러한 반응의 대부분은 토피라메이트 중단 후 가역적이었다. 만일 이 약 치료 중 어느 때라도 시각 문제가 발생한다면 약물 중단을 고려해야 한다.
4) 기분 장애 및 수면 장애
이 약은 우울증과 불안, 불면증 등 기분 장애를 유발할 수 있다. 우울증 병력이 있는 환자는 이 약을 복용하는 동안 우울증 또는 기타 기분 장애의 재발 위험이 높아질 수 있다. 이 기분 장애와 수면 장애의 대부분은 자연적으로 치료되었거나 복용 중단 시 치료되었다. 임상적으로 유의미하거나 지속적인 증상이 나타나는 경우, 이 약의 복용량을 줄이거나 복용 중단을 고려해야 한다. 환자에게서 자살 생각이나 자살 행동 증상을 보이면 이 약의 복용을 중단해야 한다.
5) 인지 장애
이 약은 인지 기능 장애(예: 주의/집중력 장애, 기억곤란, 언어 장애, 특히 단어 선택의 어려움)을 유발할 수 있다.
이 약의 용량을 빠르게 적정하거나 초기에 고용량으로 복용하면 주의력, 기억력, 언어/단어 선택 어려움과 같은 인지기능 관련 이상반응 발생률을 높이는 경우가 있다.
이 약은 인지 기능을 손상시킬 가능성이 있기 때문에, 환자는 이 약의 치료가 악영향을 미치지 않는다고 확신할 수 있을 때까지 자동차를 포함한 위험한 기계를 조작할 때 주의하여야 한다. 인지 기능 장애가 지속되면 감량을 고려해야 하며, 중등도에서 중증까지의 증상, 불편함 또는 복용량 감소로 해결하지 못한 증상에 대해 이 약의 복용량 감소 또는 복용 중단을 고려해야 한다.
6) 대사성산증
이 약으로 치료한 환자에서 음이온 차의 변화 없는(non‑anion gap) 고염소혈증성 대사성 산증(만성 호흡성 알칼리혈증 없이 정상 범위 이하로 혈청 중탄산염 농도 저하)이 보고되었다. 신장질환, 심한 호흡기질환, 간질 중첩증, 설사, 수술, 케톤 식이 등 산증의 위험을 증가시킬 수 있는 상태 혹은 치료가 토피라메이트의 중탄산염 농도 저하 효과를 증가시킬 수 있다. 이 약과 탄산탈수효소억제제(예: 조니사미드, 아세타졸아미드 또는 디클로펜아미드)를 병용할 경우 대사성산증의 중증도를 증가시킬 수 있으며 또한 신장 결석 형성의 위험도도 높아질 수 있다. 따라서 대사성산증이 발생하기 쉬운 상태의 환자가 이 약을 다른 탄산탈수효소억제제와 병용하는 경우에는 대사성산증의 발생이나 악화 여부를 모니터링 하여야 한다.
급성 또는 만성 대사성산증의 일부 증상으로는 과다호흡, 피로 및 거식증과 같은 비특이적 증상, 또는 심장부정맥 또는 혼미를 포함한 보다 심각한 후유증이 있을 수 있다. 만성적이고 치료되지 않은 대사성산증은 신석증이나 신장석회증의 위험도를 높일 수 있으며, 골연화증(소아 환자에서 구루병이라고도 함) 및/또는 골절 위험이 높은 골다공증을 유발할 수 있다. 성장 및 뼈 관련 후유증에 대한 이 약의 효과는 장기적 위약 대조 임상 시험에서 체계적으로 조사되지 않았다.
이 약의 치료 전과 치료 중에 혈청 중탄산염을 포함한 전해질 측정이 권장된다. 이 약의 임상시험에서 혈청 중탄산염의 피크 감소는 4주째까지 발생했으며, 대부분 피험자에서는 시험약 변경 없이 56주째까지 중탄산염 교정이 있었다. 그러나 이 약의 복용 중 지속적인 대사산증이 발생하면 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
7) 크레아티닌 상승
이 약은 혈청 크레아티닌 증가를 유발할 수 있다. 4주에서 8주간의 치료 후에 혈청 크레아티닌의 피크 증가가 관찰되었다. 평균적으로 혈청 크레아티닌은 점차 감소했지만 기저치 크레아티닌 값보다 높게 유지되었다. 혈청 크레아티닌의 변화(측정된 GRF)는 치료를 중단하면 회복되지만, 만성적으로 치료시에 신장 기능에 미치는 영향에 대해서는 알려지지 않았다. 따라서 이 약 치료 시작 전과 치료 중에 혈청 크레아티닌 측정이 권장된다. 이 약의 복용 중에 크레아티닌의 지속적인 상승이 발생하면 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
8) 당뇨병 치료를 하는 제2형 당뇨 환자에서 저혈당증의 잠재적 위험
체중 감량은 인슐린 및/또는 인슐린 분비 촉진제(예: 설폰요소제)로 치료되는 제2형 당뇨 환자의 저혈당증 위험을 높일 수 있다. 이 약은 인슐린과의 병용 치료에 관해서는 연구되지 않았다. 제2형 당뇨 환자에 대해서는 이 약의 치료 시작 전과 치료 중에 혈당 수치를 측정할 것을 권장한다. 저혈당의 위험을 감소시키기 위해서 비포도당 의존성(non‑glucose‑dependent) 항당뇨병제의 약물 복용량 감소를 고려해야 한다. 이 약의 복용을 시작한 후 환자가 저혈당증을 일으키면 당뇨병치료제 처방을 적절히 변경해야 한다. 또한, 이 약 투여 시 당뇨병 환자에서 인슐린 요구량이 변경될 수 있으므로 당뇨병 환자에게 식이요법과 병행하여 이 약을 투여할 경우 인슐린 투여량을 조절하여야 한다.
9) 혈압강하제로 치료받는 환자에게서 저혈압의 잠재적 위험
혈압강하제로 치료를 받는 고혈압 환자의 경우, 체중 감량은 저혈압의 위험을 높일 수 있으며 어지러움, 어질어질함, 실신 등을 유발할 수 있다. 고혈압 치료를 받는 환자는 이 약의 치료 시작 전과 치료 중 혈압 측정을 권장한다. 이 약의 복용을 시작한 후 환자에게서 저혈압과 관련된 증상을 보이면 항고혈압제 처방을 적절하게 변경해야 한다.
10) 알코올을 포함한 중추신경 억제제로 인한 중추신경기능저하
펜터민 또는 토피라메이트와 알코올 또는 중추신경 억제제(예: 바르비탈계, 벤조디아제핀 및 수면제)를 병용하면 어지러움, 인지기능 관련 이상반응, 나른함, 어질어질함, 조정력 저하 및 졸림과 같은 중추신경기능저하나 이러한 약제의 기타 중추 매개 효과를 증가시킬 수 있다. 따라서 이 약은 알코올을 함께 복용하면 안 된다.
11) 갑작스런 복용 중단으로 인한 잠재적 발작
이 약의 성분인 토피라메이트의 갑작스런 복용 중단으로 인한 금단 현상은 발작이나 간질 병력이 없는 개인에게도 발작 증상을 일으키는 경우가 있다. 이 약의 즉각적인 복용 중단이 의학적으로 요구되는 상황에서는 환자에 대한 적절한 모니터링이 권장된다. 이 약 15mg/92mg 용량의 복용을 중단하는 환자는 발작을 일으킬 가능성을 줄이기 위해 권장하는 대로 그 복용량을 점차적으로 줄여야 한다.
12) 신장애 환자
이 약의 성분인 펜터민과 토피라메이트는 신장 배설을 통해 제거된다. 따라서 중등증(크레아티닌 청소율(CrCl)이 30 mL/min 이상 50 mL/min 미만) 또는 중증(CrCl 30 mL/min 미만) 신장애 환자에서 펜터민과 토피라메이트에 대한 노출이 더 높다. 두 환자군에서 모두 이 약의 용량을 조절한다.
이 약은 신장 투석 중에 있는 말기 신장질환 환자에 대해서는 연구되지 않았다. 이 환자군에서는 이 약의 사용을 피해야 한다.
13) 간장애 환자
경증(Child‑Pugh score 5 ‑ 6) 또는 중등증(Child‑Pugh score 7 ‑ 9) 간장애 환자에서 펜터민에 대한 노출은 건강한 지원자에 비해 더 높았다. 중등증의 간장애 환자에서는 이 약의 용량을 조정한다.
이 약은 중증 간장애 환자(Child‑Pugh score 10 ‑ 15)에 대해서는 연구되지 않았다. 이 환자군에서는 이 약의 사용을 피해야 한다.
14) 신장 결석
이 약의 복용은 신장결석 형성과 관련이 있다. 이 약의 성분인 토피라메이트는 탄산탈수효소활성(carbonic anhydrase activity) 을 억제하고 뇨중 구연산의 배설을 감소시켜, 뇨의 pH를 높임으로써 신장 결석 형성을 촉진한다.
탄산탈수효소(예: 조니사미드, 아세타졸아미드, 메타졸아미드) 를 억제하는 다른 약물과 함께 이 약을 사용하지 않는다.
케톤식이요법(Ketogenic Diet)을 하는 환자의 토피라메이트 사용은 또한 신장 결석 형성의 가능성을 증가시키는 생리적 환경을 초래할 수 있다.
수분 섭취량을 늘려서 소변량을 늘리면 신장 결석 형성에 관련있는 물질의 농도를 낮출 수 있다. 운동이나 높은 기온에 노출되기 전 및 중에 적절한 수분 공급을 하는 것은 열과 관련된 이상반응의 위험을 줄일 수 있다
15) 땀분비 감소증 및 고체온증
이 약의 성분인 토피라메이트의 복용으로 입원이 필요할 수도 있는 땀분비 감소증이 보고되었다. 땀분비 감소와 정상보다 높은 체온 증가가 특징으로 나타난다. 일부 사례는 온도가 상승한 환경에 노출된 이후에 토피라메이트 복용과 함께 보고되었다.
이 약으로 치료받는 환자는 신체 활동 중, 특히 더운 날씨에 땀분비 감소와 체온 증가를 모니터링 해야 한다. 이 약을 열 관련 질환에 걸리기 쉬운 환자에게 다른 약제와 함께 처방할 때는 각별히 주의해야 한다. 이들 약제에는 다른 탄산탈수효소억제제 및 항콜린성 작용 약제가 포함되나 이에 한정되지는 않는다.
16) 저칼륨혈증
이 약은 탄산탈수효소활성의 억제를 통해 저칼륨혈증의 위험을 높일 수 있다. 또한 이 약을 푸로세미드(루프이뇨제) 또는 히드로클로로티아지드 (티아지드 유사 이뇨제)와 같은 비‑칼륨보존이뇨제(non‑potassium sparing diuretics)와 병용시 칼륨 손실을 더욱 증가시킬 수 있다. 이 약 처방 시 환자는 저칼륨혈증에 대해 모니터링되어야 한다.
17) 모니터링: 실험실적 검사
이 약은 무작위, 이중 맹검, 위약 대조 임상연구에서 여러 실험실적 분석 결과의 변화를 일으키는 경우가 있었다.
기저시점 및 치료 기간 중 주기적으로 중탄산염, 크레아티닌, 칼륨 및 혈당을 포함하는 혈액 화학 프로필을 확보해야 한다.
18) 남용
이 약의 주성분인 펜터민은 약물 남용의 가능성이 있다. 펜터민은 암페타민과 화학적 및 약리학적으로 관련이 있다. 암페타민과 기타 각성제는 광범위하게 남용되어 왔으며, 이 약을 체중 감량 프로그램의 일부로 포함시키는 것이 바람직하다고 평가할 때 펜터민의 남용 가능성을 염두에 두어야 한다. 암페타민 및 관련 약물(예: 펜터민)의 남용은 약물 사용 조절 장애와 심각한 사회적 기능 장애를 일으키는 경우가 있다. 이 약의 복용량을 권장 용량 보다 여러 번 늘린 환자에 대한 보고가 있다.
19) 의존성
이 약은 육체적 약물 의존 가능성에 대해 체계적으로 연구되지는 않았다. 육체적 약물 의존성은 반복되는 약물 복용에 대해서 생리적으로 적응하려는 상태이다. 육체적 약물 의존성은 약물 사용의 갑작스런 중단 또는 약물의 현저한 (복)용량 감소 이후에 약물 종류별 금단 현상으로 나타난다.
이 약의 각 주성분에 대한 육체적 약물 의존 가능성에 대한 정보는 한정되어 있다. 토피라메이트의 경우, 갑작스러운 약물 중단은 발작이나 간질 병력이 없는 환자에서도 발작이 일어나는 경우가 있었다. 펜터민의 경우, 장기적인 고용량 복용 후 갑작스런 중단은 극심한 피로와 정신적 우울증을 야기한다. 수면의 뇌파에도 변화가 나타난다. 따라서 이 약의 신속한 복용 중단이 필요한 상황에서는 적절한 의학적 모니터링이 권장된다.
20) 이 약은 최근 1년 이내에 다른 식욕억제제를 사용한 환자에게는 투여가 권장되지 않으므로 주의한다.
21) 토피라메이트를 투여 받은 환자에서 뇌병증을 동반 혹은 동반하지 않은 고암모니아혈증이 보고되었다. 고암모니아혈증은 토피라메이트와 발프로산을 병용 투여한 경우 더 빈번하게 보고되었다(상호작용 항 참조). 고암모니아혈증은 임상적으로 증상이 없을 수 있으나, 고암모니아혈증성 뇌병증은 기면 또는 구토를 동반한 의식수준 및/또는 인지기능에 있어서 급격한 변화를 자주 보인다. 이 약과 관련하여 설명할 수 없는 기면, 구토 혹은 정신상태의 변화가 발생한 환자의 경우, 고암모니아혈증성 뇌병증을 고려하여 혈중 암모니아 수치를 측정하는 것이 권장된다.
6. 상호작용
1) 모노아민산화효소억제제
고혈압 발생의 위험성 때문에 모노아민 산화효소 억제제(monoamine oxidase inhibitors) 투여기간 동안 또는 투여 후 14일 동안 펜터민 복용을 금한다.
2) 경구피임약
건강한 지원자와는 달리 비만환자에게서 에티닐에스트라디올(에스트로겐 성분) 35 ug과 노르에틴드론 (프로게스틴 성분) 1 mg을 함유한 경구 피임약의 단회 용량과 이 약 15 mg/92 mg의 1일 1회 병용 투여는 에티닐에스트라디올의 노출을 16% 감소시키고, 노르에틴드론의 노출을 22% 증가시켰다.
이 연구가 피임 효과에 대한 상호 작용의 영향을 구체적으로 다루지는 않았지만, 임신의 위험도가 증가할 것이라고 예상되지는 않는다. 피임 효과의 주된 결정 요인은 복합 경구 피임약의 프로게스틴 성분이므로 높아진 프로게스틴 노출이 유해할 것으로 예상되지는 않는다.
그러나 불규칙한 출혈(점상출혈)은 자궁 내막을 안정화시키는 프로게스틴에 대한 노출 증가와 에스트로겐 노출 감소로 인해 더 자주 발생할 수 있다. 환자는 출혈이 발견되면 복합 경구 피임약을 중단하지 말아야 한다는 사실을 통보 받아야 한다. 그러나 점상출혈이 불편할 경우 의사에게 알려야 한다.
3) 알코올을 포함한 중추신경억제제
이 약과 알코올 또는 기타 중추신경억제제과의 특정 약물상호작용에 관한 연구는 수행되지 않았다. 펜터민 또는 토피라메이트와 알코올 또는 중추신경억제제(예: 바르비탈류, 벤조디아제핀 및 수면제)의 병용 투여는 어지러움 또는 인지 이상반응 또는 이러한 약물의 중추 매개 효과와 같은 중추신경기능저하를 증가시킬 수 있다. 따라서 이 약을 알코올이나 다른 중추신경억제제와 함께 사용하는 경우 환자는 중추신경기능저하나 이상반응 위험이 증가할 가능성에 대해 상담받아야 한다.
4) 비‑칼륨보존이뇨제
비‑칼륨보존이뇨제와 함께 이 약을 병용투여하면 이러한 이뇨제의 칼륨 손실 작용이 증가할 수 있다. 토피라메이트 단일제와 히드로클로로티아지드 단일제를 병용하는 경우 토피라메이트의 Cmax 및 AUC가 각각 27% 및 29% 증가하는 것으로 나타났다. 비‑칼륨보존 의약품과 이 약을 병용 처방할 때, 환자의 저칼륨혈증에 대해 모니터링해야 한다.
5) 항경련제
간질 환자가 토피라메이트와 페니토인 또는 카르바마제핀을 병용하는 경우, 토피라메이트를 단독 복용하는 것에 비해 토피라메이트의 혈장 농도가 각각 48%와 40% 감소했다.
발프로산과 토피라메이트의 병용투여는 뇌병증과는 무관하게 고암모니아혈증을 일으키는 경우가 있다. 또한 발프로산과 토피라메이트를 병용한 환자에서 고암모니아혈증과 무관하게 저체온증이 나타나는 경우가 있다. 저체온증이나 뇌병증의 발병이 보고되는 경우, 환자의 혈중 암모니아 수치를 검사하는 것이 권장된다.
6) 탄산탈수효소억제제
이 약의 성분인 토피라메이트를 다른 탄산탈수효소억제제(예: 조니사미드, 아세타졸아미드,디클로펜아미드)와 함께 사용하면 대사성산증의 중증도가 증가할 수 있으며 신장 결석 형성의 위험도 증가할 수 있다. 이 약을 탄산탈수효소를 억제하는 다른 약물과 함께 사용해서는 안된다.
7) 피오글리타존
이 약의 성분인 토피라메이트와 피오글리타존의 병용투여 임상시험에서 피오글리타존과 활성대사산물 노출의 손실이 보고되었다. 이 결과의 임상적유의성은 알려지지 않았다. 그러나 피오글리타존 치료 중 이 약의 병용 또는 이 약 치료 중 피오글리타존 병용 시, 당뇨병 환자의 상태를 적절하게 통제하기 위해 환자에 대한 일상적인 모니터링 시 세심한 주의가 필요하다.
8) 아미트리프틸린
일부 환자는 이 약의 성분인 토피라메이트 복용 시 아미트리프틸린 농도가 크게 증가할 수 있다. 이 약과 병용투여 시 아미트리프틸린 용량 조절은 혈청 수치가 아닌 환자의 임상 반응에 따라 이루어져야 한다.
7. 임부 및 수유부에 대한 투여
1) 임부
이 약은 임부에게 사용해서는 안된다. 이 약은 태아에게 유해한 영향을 줄 수 있으며, 이 약 사용으로 인한 체중 감량은 임신한 여성에게 잠재적인 이득이 되지 않는다. 역학자료에서는 이 약의 성분인 토피라메이트에 임신 초기 3개월 동안 노출시 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다. 여러 종의 임신한 동물이 임상적으로 적절한 용량의 토피라메이트를 복용했을 때 두개 및 안면 결손을 포함한 구조적 기형 및 자궁 내 태아 체중 감소가 후손에서 발생했다.
환자가 임신 중에 이 약물을 복용하거나 이 약물 복용 중에 임신한 경우, 치료를 즉시 중단하고 태아에게 미칠 잠재적인 위험이 있음을 환자에게 알려야 한다.
임상적 고찰
구순구개 파열은 임신 5주부터 9주까지 발생한다. 입술은 임신 5주에서 7주 사이에 형성되며, 구개(입천장)는 임신 6주부터 9주 사이에 형성된다.
임신 중 모체 조직에서 발생하는 필수적인 체중 증가 때문에 이미 과체중이거나 비만인 임산부 뿐 아니라, 모든 임산부의 경우 최소한의 체중 증가 및 체중 감량이 되지 않는 것이 권장된다.
이 약은 대사성산증을 일으킬 수 있다. 토피라메이트 유발 대사성산증의 영향은 임신에 대해서는 연구되지 않았다. 그러나 다른 원인으로 인한 임신 중 대사성산증은 태아의 성장 감소, 태아에게 공급되는 산소 감소 및 태아 사망을 유발할 수 있으며, 분만을 견딜 수 있는 태아의 능력에 영향을 미칠 수 있다.
임상 데이터
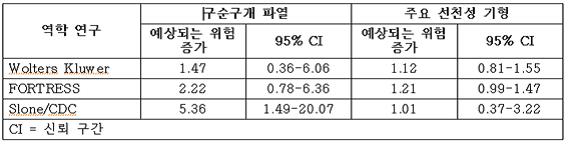
임신 중에 이 약의 구성성분인 토피라메이트 노출로 인한 주요 선천성 기형 및 구순구개 파열 위험을 평가한 데이터는 North American Anti‑Epileptic Drug (NAAED) Pregnancy Registry 및 몇 가지 대규모 후향적 역학 연구에서 확인할 수 있다. NAAED Pregnancy Registry는 구순구개 파열 위험이 9.60(95% CI 3.60 ‑ 25.70)으로 증가할 것으로 예상했다. 대규모 후향적 역학 연구에서 임신 중 토피라메이트 단일요법 노출이 구순구개 파열 위험성의 약 2~5배 증가와 연관이 있었다. (표 4). FORTRESS 연구에서는 임신 초기에 토피라메이트에 노출된 1,000명의 유아 당 구순구개 파열 증상의 위험성이 1.5배 (95% CI = ‑1.1 ~ 4.1) 증가가 관찰되었다.
표 4. 자궁 노출 및 구순구개 파열 및 주요 선천성 기형에서의 토피라메이트 연관성 평가 연구 요약
동물 데이터
펜터민/토피라메이트
태아 발육 연구는 랫드와 토끼에서 펜터민과 토피라메이트 병용 요법으로 수행되었다. 기관이 형성되는 기간에 랫드에게 투여된 펜터민과 토피라메이트는 태아의 체중 감소를 가져 왔지만, 최고용량인 펜터민 3.75 mg/kg과 토피라메이트 25 mg/kg에서 태아 기형을 일으키지 않았다. (각 주성분에 대해 AUC 예상치에 근거한 인체최대권장용량의 약 2배). 토끼를 대상으로 한 유사한 연구에서는 AUC에 근거한 인체최대권장용량의 약 0.1배(펜터민) 및 1배(토피라메이트)의 임상 노출이 있었을 때, 태아 발달에 대한 어떠한 영향도 관찰되지 않았다. 랫드와 토끼에서 이 용량으로 유의미하게 낮은 모체의 체중 증가가 기록되었다.
산전/산후 발달 연구는 펜터민과 토피라메이트 치료의 병용 요법으로 랫드에서 시행되었다. 1.5 mg/kg/day의 펜터민과 10 mg/kg/day의 토피라메이트 (AUC 기준 인체최대권장용량에서 각각 약 2배와 3배의 임상 노출)로 기관형성에서 수유기까지 치료한 랫드의 모체 또는 자손에 대한 유해한 영향은 없었다. 고용량 11.25 mg/kg/day의 펜터민과 75 mg/kg/day의 토피라메이트(AUC 기준 최대 임상 용량의 약 5배와 6배)를 투여하면 모체 체중 증가의 감소와 자손 독성을 초래한다. 자손에 대한 영향으로는 출생 후 자손 생존률 감소, 사지 및 꼬리의 기형 증가, 자손 체중 감소 및 학습, 기억 또는 수정과 생식에는 영향을 미치지 않으며 성장, 발달 및 성적 성숙을 지연시켰다. 사지 및 꼬리 기형은 토피라메이트 단독으로 수행한 동물 연구의 결과와 일치했다.
펜터민
펜터민에 대한 동물 생식 연구는 수행되지 않았다. 펜터민/토피라메이트 병용 요법으로 실시한 랫드 대상 연구로 부터 제한된 데이터는 펜터민 단독으로는 최기형성을 보이지 않았지만 AUC 기준으로 이 약의 인체최대권장용량의 5배에서 저체중과 태아의 생존률 감소를 보였다.
토피라메이트
토피라메이트는 임상적으로 적절한 용량에서 최기형성을 포함한 발달 독성을 유발한다.
2) 분만
이 약이 인간의 분만에 미치는 영향은 알려지지 않았다. 산모 및/또는 태아에서 이 약으로 유도되는 대사성산증의 발달은 분만을 견딜 수 있는 태아의 능력에 영향을 미칠 수 있다.
3) 수유부
토피라메이트와 암페타민(펜터민은 암페타민과 유사한 약리학적 활성 및 화학적 구조를 가짐)이 모유에서 배출되기 때문에 이 약이 사람의 모유에 존재할 수 있다. 신생아의 모유 수유에 대한 심각한 이상반응의 잠재적 위험성때문에, 산모에 대한 약물 처방의 중요성을 고려하여 수유를 중단할 것인지 약물 투여를 중단할 것인지에 대한 여부를 결정해야 한다.
4) 가임여성
이 약은 태아에 유해할 수 있다. 임신 등록 및 역학 연구의 자료에 따르면 임신 초기에 이 약 성분인 토피라메이트에 노출된 태아는 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다. 이 약 치료 중 임신한 여성은 즉시 치료를 중단하고 의료진에게 알려야 한다.
임신 테스트
가임기 여성은 이 약의 치료를 시작하기 전에 임신 테스트의 결과가 음성임을 확인해야 하고, 이 약으로 치료하는 동안에는 매달 한 번씩 임신 테스트를 해야 한다.
피임
이 약 치료 중 가임 여성은 효과적인 피임법을 사용해야 한다.
8. 소아에 대한 투여
18세 미만 소아 환자를 대상으로 한 이 약의 안전성과 효과는 입증되지 않았으며, 이 약의 사용은 소아 환자에게 권장되지 않는다. 이 약의 성분인 토피라메이트를 복용한 소아 환자에서 나타나는 심각한 이상반응으로는 급성 녹내장, 땀분비 감소증 및 고체온증, 대사성산증, 인지 및 신경 정신적 반응, 고암모니아혈증 및 뇌병증, 신결석이 있다.
어린 동물 연구
이 약에 대한 어린 동물 연구는 수행되지 않았다. 어린 연령(출생 후 12 ~ 50일)의 랫드에게 토피라메이트(30, 90 또는 300 mg/kg/day)를 경구 투여했을 때, 가장 높은 용량에서 수컷 랫드의 뼈 성장판 두께가 감소했다.
9. 고령자에 대한 투여
이 약의 임상 시험에서 65세 이상 환자는 총 254명(7%)이었다. 고령 피험자와 젊은 연령의 피험자 사이에 안전성이나 효과에 전반적인 차이는 없었지만, 일부 고령자에게서 나타나는 더 큰 과민반응은 배제할 수 없었다.
이 약의 임상 연구에는 젊은 연령의 피험자와 다르게 반응하는지를 판단할 수 있을 정도의 충분한 65세 이상 피험자 수가 포함되지 않았다. 일반적으로 고령 환자에 대한 투여량 선택은 신중해야 하며, 투여량 선택은 보통 투여 범위의 최하위부터 시작하며, 간, 신장 또는 심장 기능 저하와 동반 질환 또는 기타 약물 치료의 빈도가 높아지는 것을 반영해야 한다.
10. 신장애 환자
건강한 지원자와 비교할 때 C‑G 공식(Cockcroft‑Gault equation)에 의해 평가된 중등증 및 중증 신장애 환자에서 펜터민과 토피라메이트에 대해 더 높은 노출을 보였다.
경증 신장애가 있는 환자에게는 용량 조절이 필요하지 않다. 중등증(30 이상 50 mL/min 미만의 크레아티닌 청소율[CrCl]) 및 중증 (30 mL/min 미만의 CrCl) 신장애 환자에게 있어 복용량은 1일 1회 7.5 mg/46 mg을 초과해서는 안 된다.
이 약은 신장 투석 중에 있는 말기 신장질환 환자에 대해서는 연구되지 않았다. 이러한 환자군에서는 이 약의 사용을 피해야 한다.
11. 간장애 환자
경증(Child‑Pugh 5 ‑ 6) 및 중등증(Child‑Pugh 7 ‑ 9) 간장애 환자에서 펜터민에 대한 노출은 건강한 지원자에 비해 더 높았다. 이 약 성분인 토피라메이트에 대한 노출은 경증 및 중등증의 간장애 환자와 건강한 지원자 간에 유사했다.
경증의 간장애가 있는 환자에게는 용량 조절이 필요하지 않다. 중등증의 간장애가 있는 환자의 경우 1일 1회 7.5 mg/46 mg을 초과해서는 안 된다.
이 약은 중증 간장애 환자(Child‑Pugh score 10 ‑ 15)에 대해서는 연구되지 않았다. 이러한 환자군에서는 이 약의 사용을 피해야 한다.
12. 과량투여시의 처치
이 약을 과다 복용하는 경우, 과다 복용한지 얼마 되지 않았다면 즉시 위세척이나 구토를 통해 위를 비워야 한다. 환자의 임상 징후와 증상에 따라 적절한 보조요법(supportive treatment)이 제공되어야 한다.
급성 펜터민 과다 복용은 불안, 진전, 과다반사, 빠른 호흡, 착란, 공격성, 환각 그리고 공황 상태를 일으키는 경우가 있다. 피로와 우울증은 대개 중추 자극(central stimulation)에 의한다. 심혈관 영향에는 부정맥, 고혈압 또는 저혈압, 순환허탈 (circulatory collapse) 등이 있다. 위장 증상으로는 오심, 구토, 설사, 복부경련이 있다. 치명적인 중독은 보통 경련과 혼수상태로 이어진다. 식욕억제를 동반하는 만성 중독 증상은 심각한 피부병, 현저한 불면증, 과민성, 과행동증 및 성격 변화를 포함한다. 만성 중독의 심각한 징후는 정신증이며, 종종 임상적으로 정신분열증과 구별할 수 없다.
급성 펜터민 중독의 관리는 주로 증상을 관찰하는 것이며 세척과 바르비탈계 약물을 통한 진정이 포함된다. 뇨의 산성화는 펜터민 배설을 증가시킨다. 이 약의 과량 투여로 인한 급격하고 심각한 고혈압에는 가능한 신속히 펜톨아민 정맥주사하는 것이 추천된다.
토피라메이트 과다 복용으로 심각한 대사성산증이 초래되었다. 다른 징후와 증상으로는 경련, 졸림, 언어 장애, 시야흐림, 복시, 정신기능 저하, 졸음증, 협조이상, 혼미, 저혈압, 복통, 초조, 어지러움, 우울증이 있다. 대부분의 경우 임상 결과는 심각하지 않았지만, 다량의 토피라메이트를 포함한 여러 종의 약물 과다 복용 후 사망이 보고되었다. 96 ~ 110 그램의 토피라메이트를 복용한 환자는 20 ~ 24시간 지속되는 혼수상태로 입원한 다음 3 ~ 4일 후에 완전히 회복되었다.
약용탄은 시험관내에서 토피라메이트를 흡착하는 것으로 나타났다. 혈액 투석은 체내에서 토피라메이트를 제거하는 효과적인 수단이다.
13. 보관 및 취급상의 주의사항
1) 어린이의 손이 닿지 않는 곳에 보관하도록 주의한다.
2) 다른 용기에 바꾸어 넣는 것은 사고원인이 되거나 품질 유지 면에서 바람직하지 않으므로 주의한다.
14. 전문가를 위한 정보
1) 약력학적 정보
암페타민의 전형적인 작용에는 중추 신경계 자극 및 혈압 상승이 포함된다. 타키필락시스(급성 내성)와 내성은 이러한 현상이 발견된 이 부류의 모든 약물에서 입증되었다.
심장 전기생리학
무작위, 이중 맹검, 위약 대조 및 활성 대조군(목시플록사신 400 mg) 및 평행군/교차, QT/QTc 연구를 통해 QTc 간격에 대한 이 약의 영향을 평가했다. 총 54명의 건강한 피험자에게 정상 상태에서 이 약 7.5 mg/46 mg을 투여한 다음 정상 상태에서 이 약 22.5 mg/138 mg으로 적정했다. 이 약 22.5 mg/138 mg [이 약 7.5 mg/46 mg의 펜터민과 토피라메이트 최대 농도(Cmax)에 대해 각각 4배 및 3배 높은 최대치료용량] 는 QTc의 기저치로부터의 변화를 측정했을 때 심장 재분극에 영향을 미치지 않았다.
사구체여과율(GFR)
건강한 비만 남성 및 여성은 이 약(1~3일 3.75mg/23mg, 4~6일 7.5mg/46mg, 7~9일 11.25mg/69mg, 10~28일 15mg/92mg)을 4주간 복용했다. 피험자의 사구체여과율은 이오헥솔 청소율을 통해 평가되었다. 평균적으로 이 약 치료기간동안 사구체여과율은 감소하였고 이 약 복용 중단 후 4주 이내에 기저치로 회복되었다.
<BR><SPAN style="FONT‑SIZE: 11pt">2) 약물동력학적 정보</SPAN>
펜터민
이 약 단회 용량 15 mg/92mg을 경구 투여한 결과, 평균 혈장 펜터민 최대 농도(Cmax), Cmax 까지의 시간(Tmax), 투약시간부터 최종 혈중농도 정량시간 t까지의 혈중농도‑시간곡선하면적(AUC0‑t), 투약시간부터 무한시간까지의 혈중농도‑시간곡선하면적 (AUC0‑∞)은 각각 49.1ng/mL, 6시간, 1990ng▪hr/mL 및 2000ng▪hr/mL이다. 이 약 15mg/92mg에 대한 고지방 식이는 펜터민 약물동력학에 영향을 미치지 않는다. 펜터민 약물동력학은 대략 이 약 3.75mg/23mg에서 펜터민 15mg/토피라메이트 100mg까지 용량에 비례한다. 펜터민/토피라메이트 15/100mg 복합제 캡슐을 정상 상태에서 투여할 때, AUC 및 Cmax의 평균 펜터민 축적 비는 모두 약 2.5이다.
토피라메이트
이 약 단회 용량 15mg/92mg을 경구 투여한 결과, 평균 혈장 토피라메이트의 Cmax 및 Tmax, AUC0‑t 및 AUC0‑∞는 각각 1020 ng/mL, 9시간, 61600 ng▪hr/mL 및 68000 ng▪hr/mL이었다. 이 약 15mg/92mg에 대한 고지방 식이는 토피라메이트 약물동력학에 영향을 미치지 않는다. 토피라메이트 약물동력학은 대략 이 약 3.75mg/23mg에서 펜터민 15mg/토피라메이트 100mg까지 용량에 비례한다. 펜터민 15mg/토피라메이트 100mg 투여량의 복합제 캡슐을 정상 상태에서 투여할 때, AUC 및 Cmax의 평균 토피라메이트 축적 비는 모두 약 4.0이다.
분포
펜터민
펜터민은 17.5%의 혈장 단백질과 결합한다. 예상되는 펜터민 분포 용적(Vd/F)은 개체군 약물동력학적 분석 결과 348 L이다.
토피라메이트
토피라메이트는 0.5~250μg/mL 혈중농도범위에서 15‑41% 혈장 단백질과 결합한다. 혈중 토피라메이트가 증가함에 따라 결합 분획(fraction bound)이 감소했다. 개체군 약물동력학적 분석을 통해 추정된 토피라메이트 Vc/F(중앙 구획의 용적) 및 Vp/F(말초 구획의 용적)는 각각 50.8 L 및 13.1 L이다.
신진 대사 및 배설
펜터민
펜터민은 두 가지 대사경로, 즉 방향족 고리(aromatic ring)에 p‑hydroxylation과 지방족 측쇄(aliphatic side chain)에 N‑oxidation을 가지고 있다. Cytochrome P450(CYP) 3A4는 주로 펜터민을 대사하지만 광범위한 신진 대사를 보이지는 않는다. Monoamine oxidase(MAO)‑A와 MAO‑B는 펜터민을 대사하지 않는다. 단독 투여 시 뇨에서 대사되지 않은 70 ~ 80%의 펜터민이 존재한다. 평균 펜터민의 말단 반감기는 약 20시간이다. 개체군 약물동력학적 분석을 통해 예상되는 펜터민 경구 청소율(CL/F)는 8.79L/h이다.
토피라메이트
토피라메이트는 광범위한 신진 대사를 보이지 않는다. 6개의 토피라메이트 대사물질(하이드록실화(hydroxylation), 가수분해(hydrolysis) 및 글루크론산화 (glucuronidation)을 통한)이 존재하며, 그 중 어떤 것도 투여된 용량의 5% 이상을 차지하지 않는다. 단독투여시 뇨에서 대사되지 않은 70% 용량의 토피라메이트가 존재한다. 평균 토피라메이트의 말단 반감기는 약 65시간이다. 개체군 약물동력학적 분석을 통해 예상되는 토피라메이트 CL/F는 1.17L/h이다.
특정환자집단
신장애
정상적인 신장 기능을 가진 건강한 지원자와 비교하여 만성 신장애 정도가 다양한 환자에서 이 약 15 mg/92 mg의 약물동력학을 평가하기 위해 단회투여, 공개 임상시험이 수행되었다. 이 연구에는 크레아티닌 청소율을 기준으로 경증(50 이상 80 mL/min 미만), 중등증(30 이상 50 mL/min 미만) 및 중증(30 mL/min 미만)으로 분류된 신장애 환자가 포함되어 있다. 크레아티닌 청소율은 C‑G 공식(Cockcroft‑Gault equation)에 기초하여 혈청 크레아티닌으로부터 추정하였다.
건강한 지원자와 비교했을 때, 펜터민의 AUC0‑inf는 중증, 중등증 및 경증의 신장애 환자에서 각각 91%, 45%, 22%였다. 펜터민 Cmax는 2 ~ 15% 더 높았다. 건강한 지원자와 비교했을 때, 토피라메이트 AUC0‑inf는 중증, 중등증 및 경증의 신장애 환자에서 각각 126%, 85%, 25%였다. 토피라메이트 Cmax는 6 ~ 17% 더 높았다. 펜터민 또는 토피라메이트 Cmax 또는 AUC와 크레아티닌 청소율 사이의 반비례 관계가 관찰되었다. 이 약은 신장 투석 중인 말기 신장질환 환자에서는 연구되지 않았다.
간장애
경증(Child‑Pugh score 5 ‑ 6)와 중등증(Child‑Pugh score 7 ‑ 9)의 간장애 환자에 비해 정상 간 기능을 가진 건강한 지원자에서의 이 약 15 mg/92 mg의 약물동력학을 평가하기 위해 단회투여, 공개 임상시험이 수행되었다. 경증 및 중등증의 간장애 환자에서, 펜터민 AUC는 건강한 지원자와 비교하여 37% 및 60% 더 높았다. 토피라메이트의 약물동력학은 건강한 지원자와 비교할 때 경증 및 중등증의 간장애 환자에서는 영향을 받지 않는다. 이 약은 중증 간장애 환자(Child‑Pugh score 10‑15)에서는 연구되지 않았다.
약물상호작용
약물상호작용의 시험관내 평가
펜터민
펜터민은 CYP 동종 효소 CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 및 CYP3A4의 억제제가 아니며 모노아민 산화 효소의 억제제도 아니다. 펜터민은 CYP1A2, CYP2B6 및 CYP3A4의 유도 인자가 아니다. 펜터민은 P‑당단백질(P‑glycoprotein) 기질이 아니다.
토피라메이트
토피라메이트는 CYP 동종 효소 CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2D6, CYP2E1 및 CYP3A4/5의 억제제가 아니다. 그러나, 토피라메이트는 CYP2C19의 경도(mild) 억제제이다. 토피라메이트는 CYP3A4의 경도(mild) 유도 인자이다. 토피라메이트는 P‑당단백질(P‑glycoprotein) 기질이 아니다.
다른 약물에 미치는 펜터민/토피라메이트의 영향
표 6. 펜터민/토피라메이트가 병용투여 약물의 약물동력학에 미치는 영향
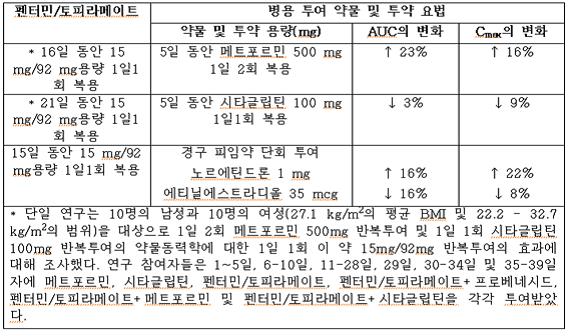
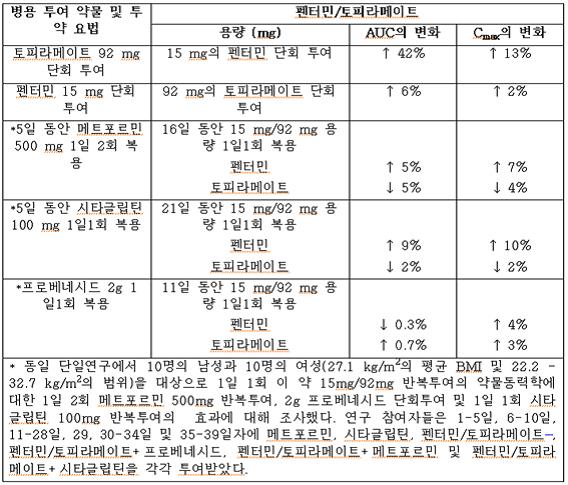
표 7. 병용투여 약물이 펜터민/토피라메이트의 약물동력학에 미치는 영향
항경련제
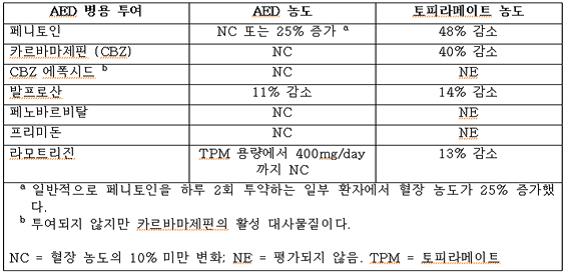
간질 환자를 대상으로 한 대조 임상 약물동력학 연구에서 토피라메이트와 표준 항경련제(AED) 간의 잠재적인 상호 작용을 평가했다. 이러한 상호 작용이 평균 혈장 AUC에 미치는 영향을 표 7에 요약했다.
표 8에서 두 번째 열(AED 농도)은 토피라메이트가 추가될 때 첫 번째 열에 나열된 항경련제(AED) 농도에 어떤 현상이 발생하는지를 설명한다. 세 번째 열(토피라메이트 농도)은 토피라메이트 단독 투여 시 실험 환경에서의 토피라메이트 농도를 첫 번째 열에 기재된 약물의 병용투여가 어떻게 변화시키는지를 설명한다.
표 8. 토피라메이트와의 항경련제(AED) 상호 작용 요약
단회 투여 연구에서 토피라메이트의 병용투여로 혈장농도곡선 아래 혈청 디곡신 면적(AUC)이 12% 감소되었다. 이러한 관찰결과에 대한 임상적 관련성은 확립되지 않았다.
히드로클로로티아지드(HCTZ)
건강한 지원자를 대상으로 실시한 약물 상호작용 연구는 정상 상태에서의 히드로클로로티아지드(25 mg 매24시간마다) 및 토피라메이트(96 mg 매12시간마다) 단독 및 병용 투여 후 약물동력학을 평가하였다.
이 연구 결과 HCTZ을 추가하였을 때 토피라메이트의 Cmax가 27%, AUC가 29% 증가되었다. 이러한 변화의 임상적 유의성은 알려져 있지 않다. 정상 상태에서의 HCTZ의 약물동력학은 토피라메이트의 병용으로 유의한 영향을 받지 않았다. 토피라메이트와 HCTZ 투여 후 혈청 칼륨의 저하가 관찰되었으며 두 약물을 병용 투여했을 때 더 크게 나타났다.
피오글리타존
건강한 자원자를 대상으로 한 약물상호작용 연구에서 7일 동안 토피라메이트(96mg 1일 2회)와 피오글리타존(30mg 1일)을 단독 및 병용 투여 후 정상상태에서의 약물동력학을 평가하였다. 피오글리타존의 AUCt,ss가 15% 저하되었으며 Cmax,ss에는 변화가 관찰되지 않았다. 이 결과는 통계적으로 유의하지 않았다. 활성 수산화‑대사체의 Cmax,ss와 AUCt,ss는 각각 13%, 16% 저하되었으며, 활성 keto‑대사체의 Cmax,ss와 AUCt,ss는 60% 저하되었다. 이 결과의 임상적 유의성은 알려져 있지 않다. 피오글리타존 치료를 받는 환자에 이 약을 투여하거나 이 약을 투여받는 환자에 피오글리타존을 투여할 때 당뇨가 적절히 조절되고 있는지 모니터하는데 주의한다.
글리부리드
제2형 당뇨병 환자를 대상으로 한 약물상호작용 연구에서 글리부리드 단독(5mg/day) 및 토피라메이트와 병용(150mg/day)시의 정상상태 약물동력학을 평가하였다. 토피라메이트 투여 중 Cmax는 22% 감소하였고 글리부리드 AUC24가 25% 감소하였다. 또한, 활성 대사체 4‑trans‑히드록시글리부리드(M1) 및 3‑cis‑히드록시글리부리드(M2)의 전신노출은 각각 13% 및 15%까지 저하되었고, Cmax는 각각 18%와 25% 감소했다. 토피라메이트의 정상상태 약물동력학은 글리부리드의 병용투여에 영향을 받지 않았다.
리튬
토피라메이트 200 mg/day 투여 중 환자에서 리튬의 약물동력학은 영향을 받지 않았다. 그러나 토피라메이트를 600 mg/day까지 투여한 후 리튬의 전신 노출이 증가하는 것으로 나타났다(Cmax는 27%, AUC는 26%). 고농도의 토피라메이트와 병용 투여시 리튬 수치를 모니터링해야 한다.
할로페리돌
할로페리돌(5mg) 단회 투여의 약물동력학은 13명의 건강한 성인(남성 6명, 여성 7명)에서 토피라메이트(12시간마다 100mg)의 반복 투여에 의한 영향을 받지 않았다.
아미트립틸린
토피라메이트 200 mg/day를 투여받은 일반인 18명(남성 9명, 여성 9명)에서 아미트립틸린(하루 25mg)의 AUC와 Cmax가 12% 증가했다. 일부 피험자에서 토피라메이트를 복용 시 아미트립틸린 농도가 크게 증가할 수 있으며, 아미트립틸린 용량 조절은 혈청 수치가 아닌 환자의 임상 반응에 따라 이루어져야 한다.
수마트립탄
24명의 건강한 지원자(남성 14명, 여성 10명)에서 토피라메이트(12시간마다 100mg)의 반복 투여는 단회 투여의 수마트립탄의 경구(100mg) 또는 피하 투여(6mg)의 약물동력학에 영향을 미치지 않았다.
리스페리돈
토피라메이트를 100, 250, 400mg/day 로 용량을 증가하여 병용투여시 리스페리돈의 전신 노출이 감소되었다(250, 400mg/day에서 각각 정상상태 AUC의 16%, 33% 감소). 9‑히드록시히스페리돈 수치의 변화는 관찰되지 않았다. 리스페리돈과 토피라메이트 400 mg/day 병용투여 시 토피라메이트의 Cmax가 14% 증가하였고, AUC12가 12% 증가했다. 리스페리돈과 9‑히드록시히스페리돈 또는 토피라메이트의 전신 노출에 대해 임상적으로 유의한 변화는 없었으므로, 이 상호작용은 임상적으로 유의하지 않을 것으로 예상된다.
프로프라놀롤
34명의 건강한 지원자(남성 17명, 여성 17명)에 대한 토피라메이트(200mg/day) 반복 투여는 일일 160mg 용량의 프로프라놀롤 약물동력학에 영향을 미치지 않았다. 39명의 지원자(남성 27명, 여성 12명)에서 160mg/day의 프로프라놀롤 복용량은 200mg/day 용량의 토피라메이트 노출에 아무런 영향을 미치지 않았다.
디히드로에르고타민
24명의 건강한 지원자(남성 12명, 여성 12명)에서 토피라메이트(200mg/day)의 반복 투여는 1mg 피하 투여량의 디히드로에르고타민 약물동력학에 영향을 미치지 않았다. 유사하게, 디히드로에르고타민의 1 mg 피하 투여량은 동일한 연구에서 토피라메이트의 200 mg/day를 투여했을 때의 약물동력학에 영향을 미치지 않았다.
딜티아젬
토피라메이트(150mg/day)와 딜티아젬(240mg)을 병용투여 시, 딜티아젬 Cmax가 10% 감소하고 AUC가 25% 감소했으며, des‑acetyl diltiazem의 AUC는 18%, Cmax가 27% 감소하고, N‑desmethyl diltiazem에는 영향을 미치지 않았다. 토피라메이트와 딜티아젬의 병용투여는 결과적으로 토피라메이트의 Cmax를 16% 증가시키고, AUC12를 19% 증가시킨다.
벤라팍신
건강한 지원자에서 토피라메이트(150mg/day)를 반복투여 시 벤라팍신 또는 O‑desmethyl 벤라팍신의 약물동력학에 영향을 미치지 않았다. 벤라팍신(150 mg 서방제제)의 반복 투여는 토피라메이트의 약물동력학에 영향을 미치지 않았다.
3) 독성시험 정보
⓵ 발암성, 변이원성, 수태능이상
펜터민/토피라메이트
펜터민/토피라메이트 복합제로 발암성, 변이원성 또는 수태능이상을 평가한 동물 실험은 없었다. 다음 데이터는 이 약의 두 가지 주성분인 펜터민 또는 토피라메이트로 개별적으로 수행한 연구 결과를 바탕으로 한다.
펜터민
펜터민은 Ames 박테리아 변이원성 시험, Chinese 햄스터 폐 세포로 시행한 염색체 이상 분석 또는 생체내 소핵 분석에서 대사 활성화 또는 대사 증진이 없는 변이원성 또는 염색체성 물질이 아니다.
랫드에게 3, 10, 30 mg/kg/day의 펜터민을 2년간 경구 투여하였다. AUC 노출에 근거하여, 이 약 15mg/92mg의 최대 권장 투여량의 약 11~15배인 최고 용량의 펜터민(30 mg/kg)에서 발암성에 대한 증거는 없었다.
수태능이상에 대한 잠재력을 결정하기 위해 펜터민으로 수행한 동물실험은 없었다.
토피라메이트
토피라메이트는 일련의 시험관내/생체내 분석 시험에서 유전 독성 잠재력을 보이지 않았다. 토피라메이트는 Ames 시험이나 시험관내 마우스 림프종 시험에서 돌연변이성을 갖지 않았다. 그것은 시험관내에서 랫드의 간세포에서 부정기 DNA 합성(unscheduled DNA synthesis)을 증가시키지 않았으며, 시험관 내 인간 림프구나 생체 내 랫드의 골수에서 염색체 이상을 증가시키지 않았다.
21개월 동안 식사를 통해 토피라메이트(20, 75 및 300 mg/kg)를 투여한 마우스에서 방광 종양의 증가가 관찰되었다. 300 mg/kg을 투여 받은 암컷과 수컷에서 통계적으로 유의한 방광 종양 발생률의 상승은 주로 마우스에 조직 형태학적으로 고유한 평활근 종양의 증가로 인한 것이다. 300 mg/kg을 투여 받은 마우스의 혈장 노출은 이 약 15 mg/92 mg의 인체최대권장용량인 토피라메이트 단일요법을 받은 환자에서 측정한 정상 상태 노출의 약 2 ~ 4배이었다. 인체 발암성에 대한 이러한 발견의 관련성은 아직은 불확실하다. 최대 120 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 약 4 ~ 10배)까지의 용량으로 2년간 토피라메이트를 경구 투여한 결과 랫드에서 발암성의 증거는 발견되지 않았다.
최대 100mg/kg 또는 AUC에 근거한 이 약의 남성 및 여성에 대한 인체최대권장용량의 약 4 ~ 8배 노출 시 랫드에서 암수컷의 수태능에 영향이 없었다.
⓶ 생식발생독성시험
토피라메이트
이 약 성분인 토피라메이트는 여러 동물 종에서의 임상적으로 적절한 용량에서 최기형성을 비롯한 발생 독성을 유발한다.
기관형성기에 임신한 마우스에게 20, 100, 500 mg/kg으로 경구 투여했을 때, 모든 투여량에서 태아 기형 초기 두개안면결손) 발생률이 증가했다. 이 연구에서 토피라메이트 저용량(20 mg/kg)은 이 약의 15 mg/92 mg에서 mg/m2기준으로 토피라메이트의 인체최대권장용량의 약 2배이다. 태아의 체중과 골격골형성은 모체 체중 증가의 감소와 함께 500 mg/kg으로 감소되었다.
랫드 연구 (경구 투여량 20, 100, 500 mg/kg 또는 0.2, 2.5, 30, 400 mg/kg)에서 사지기형 (손발가락결손증 (ectrodactyly), 사지단소증 (micromelia), 무지증 (amelia))의 빈도는 임신 중 기관형성기간 동안 400 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량 34배) 이상으로 치료한 암컷의 자손사이에서 증가했다. 배아독성(태아 체중 감소, 구조적 변화의 발생률 증가)은 20 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량 2배) 정도의 낮은 용량에서 관찰되었다. 모체 독성에 대한 임상적 징후는 400 mg/kg 이상에서 나타나고, 모체의 체중 증가는 100 mg/kg 혹은 그 용량 이상으로 치료하는 동안 감소했다.
토끼 연구(기관형성기간 동안 경구로 20, 60, 180 mg/kg 또는 10, 35, 120 mg/kg)에서는 배/태아 사망률이 35 mg/kg(AUC 추정치에 근거한 인체최대권장용량의 2배)이상에서 증가였고, 최기형성의 영향(주로 늑골 및 척추 기형)이 120 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 6배)에서 관찰되었다. 모체 독성(체중 증가의 감소, 임상적 증후 및/또는 사망률)의 증거가 35 mg/kg 이상에서 나타났다.
임신 후반기와 수유기(0.2, 4, 20, 100 mg/kg 또는 2, 20, 200 mg/kg)의 암컷 랫드에 투여했을 때, 자손은 200 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 16배)에서 생존력이 감소하고 신체 발달이 지연되었으며, 2 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 2배) 이상에서 이유기 전 및/또는 후에 체중 증가가 감소하는 것으로 나타났다. 모체 독성(체중 증가의 감소, 임상 징후)은 100 mg/kg 혹은 그 이상에서 나타났다.
산후 성분(기관형성 중 0.2, 2.5, 30, 400 mg/kg)을 갖은 랫드의 배아/태아 발생 연구에서, 자손은 400 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 34배)에서 신체 발달이 지연되었고, 30 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 2배)이상에서 체중 증가의 지속적인 감소가 나타났다.
⓷ 임상시험 정보
감소된 칼로리 섭취 및 늘어난 신체 활동과 관련하여 체중 감량에 대한 이 약의 효과는 2개의 무작위배정, 이중맹검, 위약 대조 임상시험에서 비만 환자군(시험 1)와 비만 혹은 2가지 이상의 유의한 동반질환을 갖는 과체중 환자군(시험 2)에서 연구되었다. 두 임상시험 모두 4주간의 적정 기간이후 52주간의 치료 기간이 있었다. 치료 1년 후(56 주)에 측정된 2가지의 공동 1차 유효성 결과지표는 1) 기저치로부터의 체중 감소 백분율과 2) 기저치로부터 최소한 5%의 체중 감소 달성으로 정의되는 치료에의 반응이었다.
시험 1에서 비만 환자군(BMI 35kg/m2이상)는 1년 동안 2:1:2의 비율로 514명은 위약을, 241명은 이 약 3.75mg/23mg을, 512명은 이 약 15mg/92mg을 무작위로 처방 받았다. 환자는 18 ~ 71세(평균 연령 43세)로 구성되었으며, 83%가 여성이었다. 약 80%는 백인, 18%는 아프리카 계 미국인, 15%는 히스패닉/라틴계였다. 임상시험 시작 당시 환자의 평균 체중과 BMI는 각각 116kg과 42kg/m2였다. 제2형 당뇨 환자는 시험 1 참여에서 제외되었다. 이 임상시험 동안에 모든 환자에게 칼로리 섭취량이 약 500 kcal/day 감소하도록 균형 잡힌 저칼로리 식이 요법을 권장했으며, 영양 및 생활습관의 변화에 관한 상담을 제공했다.
시험 2에서 과체중 및 비만 환자군은 1년 동안 2:1:2의 비율로 994명은 위약을, 498명은 이 약 7.5mg/46mg을, 995명은 이 약 15mg/92mg을 무작위로 처방받았다. 임상시험에 적합한 환자는 27kg/m2 이상 45kg/m2 이하의 BMI(제2형 당뇨 환자의 경우 BMI에 대한 하한 값은 없음)와 다음의 비만관련 합병증에서 2가지 이상의 조건을 충족해야 했다.
• 고혈압(140/90 mmHg 이상 또는 당뇨인 경우 130/85 mmHg 이상) 또는 2가지 이상의 혈압강하제가 필요한 환자
• 200‑400 mg/dL 이상의 트리글리세리드 또는 2개 이상의 지질 저하 약물로 치료를 받고 있는 환자
• 공복 혈당 상승(100 mg/dL 이상) 또는 당뇨병;
• 허리 둘레: 남성 102cm 이상, 여성 88cm 이상
환자는 19 ~ 71세(평균 연령 51세)로 구성되었으며, 70%가 여성이었다. 약 86%는 백인, 12%는 아프리카 계 미국인, 13%는 히스패닉/라틴계였다. 임상시험 시작 당시 환자의 평균 체중과 BMI는 각각 103kg과 36.6kg/m2였다. 임상시험 시작 시 환자의 약 절반(53%)이 고혈압을 앓았다. 연구 시작 당시 제2형 당뇨 환자는 388명 (16%)이었다. 이 연구 동안에 모든 환자에게 칼로리 섭취량이 약 500 kcal/day 감소하도록 균형 잡힌 저칼로리 식이 요법을 권장했으며, 환자에게 영양 및 생활습관의 변화에 관한 상담을 제공했다.
무작위 배정된 환자 중 시험 1에서 40%, 시험 2에서 31%에 해당하는 상당수의 피험자가 56주 전에 연구를 중단하였다.
표 9은 시험 1과 시험 2에서 1년간의 체중 감량 결과를 나타낸다. 이 약 치료 1년 후, 모든 용량에서 위약에 비해 통계적으로 유의한 체중 감량을 초래했다 (표 9, 그림 1 및 2). 위약군 대비 이 약에 무작위 배정된 환자들의 통계적으로 상당히 유의한 비율에서 5% 및 10%의 체중감량을 달성하였다.
표 9. 시험 1과 시험 2에서 1년간의 체중 감량
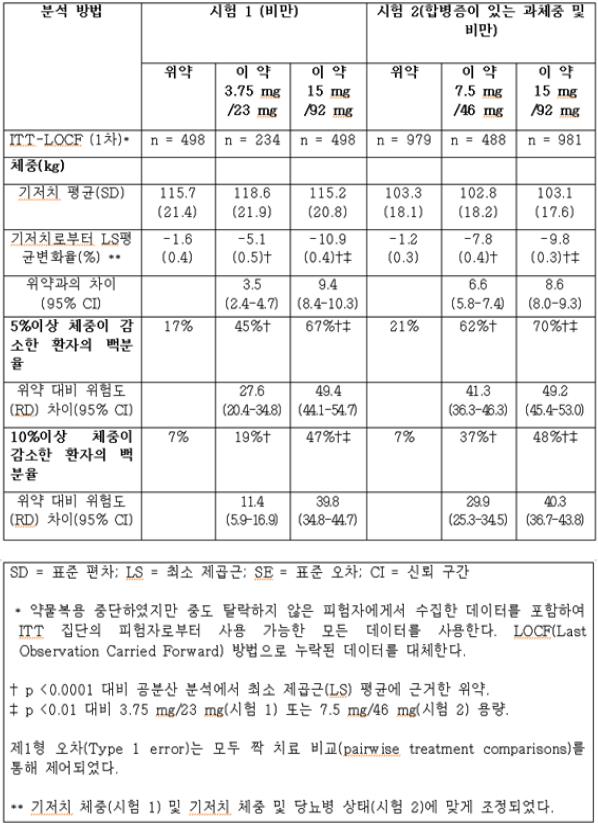
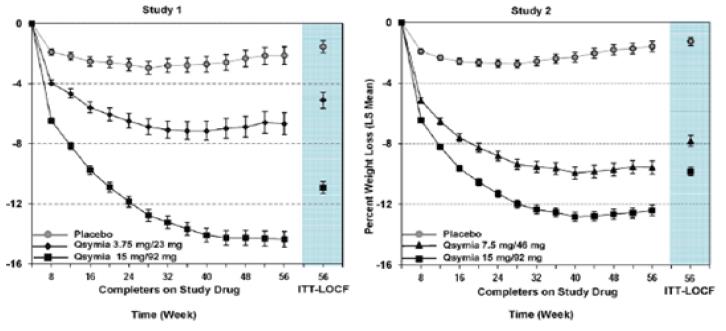
그림 1. 시험 1 체중 변화 비율 그림 2. 시험 2 체중 변화 비율
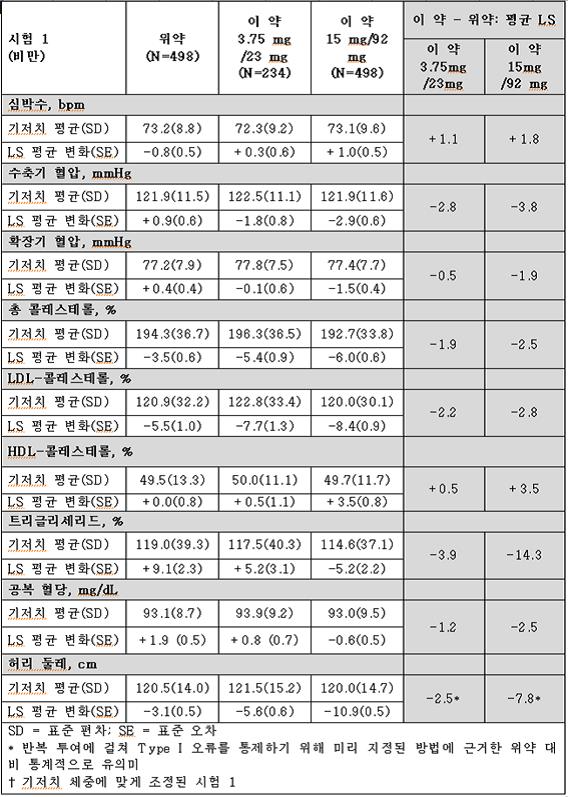
시험 1과 시험 2의 비만과 관련된 심혈관, 대사 및 인체 계측 위험 요소의 변화는 표 10와 표 11에 나타내었다.
이 약을 사용한 1년간의 치료로 심박수를 제외하고 비만과 관련된 여러 위험 요소에서 위약보다 상대적으로 호전이 있었다.
표 10. 시험 1(비만)에서 1년간의 치료 후 위험 인자에 대한 기저치로부터의 최소 제곱근 평균† 변화(Least‑Squares (LS) Mean Change) 및 위약군과의 치료 결과 차이
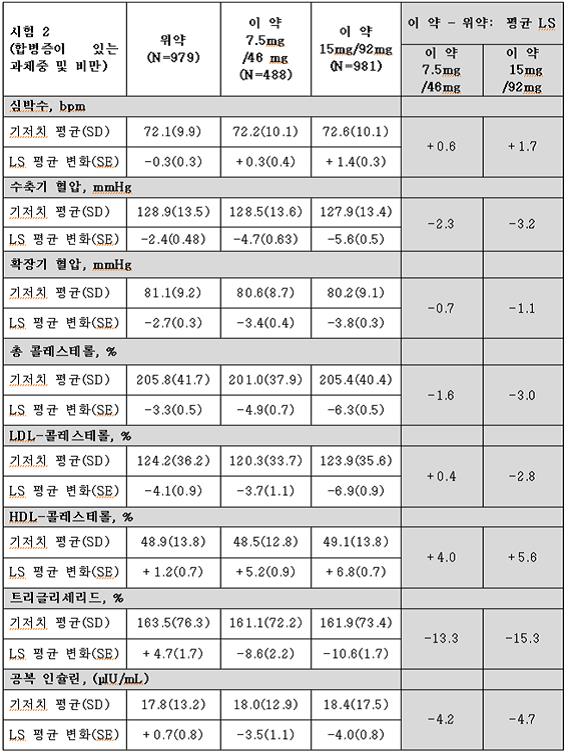
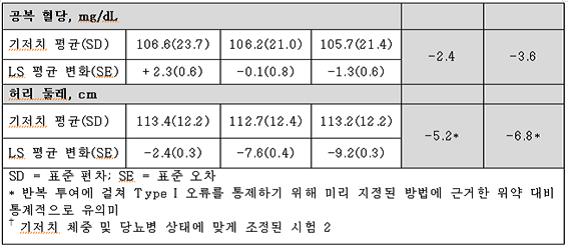
시험 2에서 치료한 제2형 당뇨가 있는 388명의 대상자 중 기저치 대비 HbA1c 감소(6.8%)는 위약군에서는 0.1% 감소한 반면, 이 약 7.5 mg/46 mg 군 및 15 mg/92 mg군에서는 각각 0.4%씩 감소했다.
다음의 초기 체질량 지수(BMI)의 성인 환자에서 저칼로리 식이 요법과 만성 체중 관리를 위한 신체 활동 증가의 보조요법으로 사용된다.
◦ 30 kg/m<SUP>2</SUP>이상 또는
◦ 고혈압, 제2형 당뇨병 또는 이상지질혈증과 같은 적어도 하나의 체중 관련 동반질환이 있는 경우 27 kg/m<SUP>2</SUP>이상
이 약은 다른 식욕억제제와 병용하지 않고 단독으로만 사용해야 한다.
◦ 30 kg/m<SUP>2</SUP>이상 또는
◦ 고혈압, 제2형 당뇨병 또는 이상지질혈증과 같은 적어도 하나의 체중 관련 동반질환이 있는 경우 27 kg/m<SUP>2</SUP>이상
이 약은 다른 식욕억제제와 병용하지 않고 단독으로만 사용해야 한다.
이 약은 음식 섭취와 무관하게 매일 아침에 복용하고, 불면증이 생길 수 있으니 저녁 복용을 피한다.
1) 초회 용량으로 이 약 3.75mg/23mg 용량을 매일 14일간 복용한다.
2) 14일 이후에는 권장량 7.5mg/46mg 용량을 12주간 복용한다.
3) 체중 감량을 확인하여 7.5mg/46mg 복용으로 최초 투여시점 전 체중 대비 3% 이상을 감량하지 못한 경우, 복용을 중단하거나 복용량을 증량한다.
4) 복용량을 증량하는 경우, 11.25mg/69mg 용량을 매일 14일간 복용한다.
5) 14일 이후에는 15mg/92mg 용량을 12주간 복용한다.
6) 체중 감량을 확인하여 15mg/92mg 복용으로 최초 투여시점 전 체중 대비 5% 이상을 감량하지 못한 경우, 복용을 중단한다.
*3.75mg/23mg 과 11.25mg/69mg 용량은 적정을 위해서만 사용한다.
복용 중단
갑작스러운 복용 중단은 발작을 일으킬 가능성이 있기 때문에 치료를 완전히 중단하기 전에 적어도 1주일 동안 하루 걸러 한 번 복용하여 점차적으로 15mg/92mg 용량의 복용을 중단한다.
신장애 환자
중등도 (30 이상 50 mL/min 미만의 크레아티닌 청소율 [CrCl]) 또는 중증 (30 mL/min 미만의 CrCl)의 신장애 환자에서는 1일 1회 7.5mg/46mg을 초과해서는 안 된다.
신장애 환자는 실제 체중과 함께 Cockcroft ‑Gault 계산식을 사용해서 CrCl 계산을 통해 결정된다.
간장애 환자
중등도의 간장애 환자(Child ‑Pugh score 7 ‑9)에서는 1일 1회 7.5mg/46mg을 초과해서는 안 된다.
1) 초회 용량으로 이 약 3.75mg/23mg 용량을 매일 14일간 복용한다.
2) 14일 이후에는 권장량 7.5mg/46mg 용량을 12주간 복용한다.
3) 체중 감량을 확인하여 7.5mg/46mg 복용으로 최초 투여시점 전 체중 대비 3% 이상을 감량하지 못한 경우, 복용을 중단하거나 복용량을 증량한다.
4) 복용량을 증량하는 경우, 11.25mg/69mg 용량을 매일 14일간 복용한다.
5) 14일 이후에는 15mg/92mg 용량을 12주간 복용한다.
6) 체중 감량을 확인하여 15mg/92mg 복용으로 최초 투여시점 전 체중 대비 5% 이상을 감량하지 못한 경우, 복용을 중단한다.
*3.75mg/23mg 과 11.25mg/69mg 용량은 적정을 위해서만 사용한다.
복용 중단
갑작스러운 복용 중단은 발작을 일으킬 가능성이 있기 때문에 치료를 완전히 중단하기 전에 적어도 1주일 동안 하루 걸러 한 번 복용하여 점차적으로 15mg/92mg 용량의 복용을 중단한다.
신장애 환자
중등도 (30 이상 50 mL/min 미만의 크레아티닌 청소율 [CrCl]) 또는 중증 (30 mL/min 미만의 CrCl)의 신장애 환자에서는 1일 1회 7.5mg/46mg을 초과해서는 안 된다.
신장애 환자는 실제 체중과 함께 Cockcroft ‑Gault 계산식을 사용해서 CrCl 계산을 통해 결정된다.
간장애 환자
중등도의 간장애 환자(Child ‑Pugh score 7 ‑9)에서는 1일 1회 7.5mg/46mg을 초과해서는 안 된다.
1. 경고
1) 이 약은 전문의약품, 일반의약품, 생약제제를 포함하여 다른 식욕억제제와 병용하여 사용하여서는 안된다. 이 약은 체중감량을 목적으로 투여하는 다른 의약품과의 병용투여에 대한 안전성▪유효성은 확립되지 않았다. 따라서, 이 약과 체중조절 목적으로 하는 다른 약물과의 병용은 권장되지 않는다.
2) 태아 독성 : 이 약은 태아에 유해한 영향을 줄 수 있다. 임신 및 역학 관련 연구 자료에 따르면 임신 초기에 이 약의 성분인 토피라메이트에 노출된 태아는 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다.
환자가 임신 중에 이 약을 복용하거나 이 약 복용 중에 임신한 경우, 치료를 즉시 중단하고 태아에게 미칠 잠재적인 위험이 있음을 환자에게 알려야 한다.
가임기 여성은 이 약으로 치료를 시작하기 전에 임신 테스트의 결과가 음성임을 확인해야 하고, 이 약으로 치료하는 동안에는 매달 한 번씩 임신 테스트를 해야 한다. 이를 위해 1회 1개월분 이상의 약을 처방하지 않아야 한다. 가임기 여성은 이 약으로 치료 중에는 효과적인 방법으로 피임을 해야 한다. [임부 및 수유부에 대한 투여 (7.1) 및 (7.4) 참조]
3) 이 약의 성분인 토피라메이트를 투여 받은 환자에서 중대한 피부반응(스티븐스존슨 증후군(SJS) 및 독성표피괴사용해(TEN))이 보고되었다. 대부분의 경우 스티븐슨존슨 증후군(SJS) 및 독성표피괴사용해(TEN)와 연관이 있다고 알려져 있는 다른 약물들을 함께 복용한 환자에서 발생하였으나, 몇몇 사례는 토피라메이트를 단독으로 복용하는 환자에서도 보고되었다. 따라서 중대한 피부반응의 징후를 환자에게 알리는 것이 권고된다. 스티븐슨존슨 증후군(SJS) 또는 독성표피괴사용해(TEN)가 의심될 경우, 이 약의 사용을 중단해야 한다.
2. 다음 환자에는 투여하지 말 것
1) 임신부
2) 녹내장 환자
3) 갑상선 기능 항진증 환자
4) MAO 억제제를 복용중이거나 또는 복용 후 14일이 경과하지 않은 환자 (혈압 상승 위험 유발)
5) 교감신경흥분성 아민에 대한 과민 반응 또는 특이체질 환자
6) 이 약의 성분에 과민증이 있는 환자
7) 18세 미만의 소아
8) 진전된 동맥경화증 환자
9) 심혈관계 질환 환자
10) 중등도 ~ 중증의 고혈압 환자
11) 폐동맥 고혈압 환자
12) 정신적으로 매우 불안하거나 흥분상태에 있는 환자
13) 약물남용의 병력이 있는 환자
3. 다음 환자에는 신중히 투여할 것
1) 신장애 환자
2) 간장애 환자
3) 이 약은 황색4호(타르트라진)를 함유하고 있으므로 이 성분에 과민하거나 알레르기 병력이 있는 환자에는 신중히 투여한다.(큐시미아캡슐7.5mg/46mg, 큐시미아캡슐11.25mg/69mg, 큐시미아캡슐15mg/92mg에 한함)
4) 이 약은 황색5호(선셋옐로우 FCF, Sunset Yellow FCF)를 함유하고 있으므로 이 성분에 과민하거나 알레르기 병력이 있는 환자에는 신중히 투여한다.(큐시미아캡슐7.5mg/46mg, 큐시미아캡슐11.25mg/69mg, 큐시미아캡슐15mg/92mg에 한함)
4. 이상반응
1) 임상시험
임상시험은 광범위하고 다양한 조건에서 시행되므로, 약물의 임상시험에서 관찰되는 이상반응 비율을 다른 약물의 임상시험에서 관찰되는 이상반응 비율과 직접적으로 비교할 수 없으며, 실제로 관찰되는 비율에 반영되지 않을 수도 있다.
이 자료는 평균 298일 동안 노출된 2,318명의 성인 환자(고혈압 환자 936명[40.4%], 제2형 당뇨 환자 309명[13.3%], BMI가 40 kg/m2 초과인 환자 808명[34.9%])를 대상으로 한 2개의 1년, 무작위, 이중 맹검, 위약 대조, 다기관 임상시험과 2개의 2상 임상 보조 연구를 통해 이 약에 대한 노출을 반영하여 기술하였다.
흔한 이상반응 : 5% 이상, 위약에 비해 최소 1.5배 이상으로 나타나는 이상반응으로는 지각 이상, 어지러움, 미각이상, 불면증, 변비, 구강 건조 등이 있다.
이 약의 치료 환자군의 2% 이상 및 위약군에서 보다 빈번하게 보고된 이상 반응은 표 1에 나와 있다.
표 1. 1년의 치료기간 동안 환자의 2% 이상 및 위약군에서 자주 보고된 이상 반응 ‑ 전반적인 연구 개체군 (Overall Study Population)
지각 이상/미각이상
이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 및 위약을 복용한 환자에서 손, 발, 얼굴 따끔거림 등의 지각 이상이 각각 4.2%, 13.7%, 19.9%, 1.9% 발생했다.
미각이상은 금속성 맛(metallic taste)으로 특징되고, 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 투여군에서 각각 1.3%, 7.4%, 9.4% 발생했으며, 이는 위약 투여군에서 1.1% 발생했다. 이러한 증상 대부분은 약물 치료 후 첫 12주 이내에 발생했다. 그러나 일부 환자군에서는 치료하는 동안 나중에 발생하는 것으로 보고되었다. 이 약 치료군에 해당하는 환자의 일부가 이러한 증상으로 인해 치료를 중단했다. (지각 이상의 경우 1%, 미각이상의 경우 0.6%).
기분 장애 및 수면 장애
이 약의 1년간 위약‑대조임상 시험에서 기분 장애 및 수면 장애와 관련된 하나 이상의 이상반응을 보고한 환자의 비율은 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 용량에서 각각 15.8%, 14.5%, 20.6%이었으며, 위약의 경우 10.3%였다. 이 증상들은 수면 장애, 불안 및 우울증으로 분류되었다. 수면 장애에 대한 보고는 일반적으로 불면증의 특징을 보였고, 이 약 3.75mg/23mg, 7.5mg/46mg 및 15mg/92mg 용량으로 치료받은 환자군에서 각각 6.7%, 8.1%, 11.1% 발생했으며, 위약으로 치료받은 환자군의 5.8%에서 발생했다. 불안은 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 용량으로 치료받은 환자군에서 각각 4.6%, 4.8%, 7.9% 발생했으며, 위약으로 치료받은 환자군의 2.6%에서 발생했다. 우울증/기분 장애에 대한 보고는 이 약 3.75mg/23mg, 7.5mg/46mg 및 15mg/92mg 용량으로 치료받은 환자군에서 각각 5.0%, 3.8% 및 7.6% 발생했으며, 위약으로 치료받은 환자군에서 3.4% 발생했다. 이러한 증상 대부분은 약물 치료 후 첫 12주 이내에 발생했다. 그러나 일부 환자군에서는 치료 과정에서 나중에 발생하는 것으로 보고되었다. 이 약의 임상 시험에서 기분과 수면 이상반응의 전반적인 유병률은 우울증 병력이 없는 환자에 비해 우울증의 병력이 있는 환자군에서 약 2배나 높았다. 그러나 기분 및 수면 이상반응이 나타난 환자들의 비율은 이 약으로 치료를 받는 환자군과 위약으로 치료를 받은 환자군에서 비슷하게 나타났다. 우울증 관련 증상은 모든 치료군에서 우울증 과거력이 있는 환자에서 더 빈번하게 발생했다. 그러나 이러한 증상의 발생률에 대한 위약‑보정된 차이(placebo‑adjusted difference)는 과거 우울증 병력에 관계없이 집단 간에 일정하게 유지되었다.
인지 장애
1년간의 위약‑대조임상 시험에서 1개 이상의 인지 관련 이상반응을 겪은 환자의 비율은 3.75mg/23mg 용량에서 2.1%, 7.5mg/46mg 용량에서 5.0%, 15mg/92mg 용량에서 7.6%, 위약에서 1.5% 였다. 이러한 이상반응은 주로 주의/집중력, 기억력, 언어(단어 선택) 문제였다. 이 증상들은 일반적으로 치료 첫 4주 이내에 시작되었고, 평균적으로 약 28일 이하의 기간 동안 지속되었으며, 치료를 중단하면 원상태로 회복되었다. 그러나 특정 환자의 경우 치료 후반 및 장기간동안 이 증상들을 경험했다.
실험실검사수치 이상
⓵ 혈청 중탄산염
1년간의 위약‑대조임상 시험에서 정상 범위 이하(2회 연속 방문 시 또는 최종 방문 시 21 mEq/L 미만의 수준)의 혈청 중탄산염의 지속적인 treatment‑emergent(TE) 감소 발생률은 3.75mg/23mg 투여군에서 8.8%, 7.5mg/46mg 투여군에서 6.4%, 15mg/92mg 투여군에서 12.8%, 위약 투여군에서 2.1%였다. 지속적이고 현저하게 낮은 혈청 중탄산염 수치(2회 연속 방문 시 또는 최종 방문 시 17 mEq/L 미만의 수준) 발생률은 3.75mg/23mg 투여군에서 1.3%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.7%, 위약 투여군에서 0.1% 였다. 일반적으로 혈청 중탄산염 농도의 감소는 경도 수준(평균 1‑3 mEq/L)이었고 치료 초기(4주 방문)에 발생했지만, 치료 후기에 중증 감소(severe decreases) 및 감소(decreases)가 발생하기도 했다.
⓶ 혈청 칼륨
1년간의 위약‑대조임상시험 중 지속적으로 낮은 혈청칼륨 수치(2회 연속 방문 시 또는 최종 방문 시 3.5 mEq/L 미만)의 발생률은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 3.6%, 15mg/92mg 투여군에서 4.9%, 위약 투여군에서 1.1%였다. 지속적으로 낮은 혈청칼륨(low serum potassium)을 경험한 피험자들 중 88%는 비‑칼륨보존이뇨제 (non‑potassium sparing diuretic)로 치료를 받았다.
임상시험기간 동안 현저히 낮은 혈청칼륨(3 mEq/L 미만, 치료 전보다 0.5 mEq/L 이상 감소)의 발생률은 3.75mg/23mg 투여군에서 0.0%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.7%, 위약 투여군에서 0.0%였다. 지속적으로 현저히 낮은 혈청칼륨(3 mEq/L 미만 및 2회 연속 방문시 또는 최종 방문시 치료 전보다 0.5 mEq/L 초과 감소)의 발생률은 3.75mg/23mg 투여군에서 0.0%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.1%, 위약 투여군에서 0.0%였다.
저칼륨혈증은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 1.4%, 15mg/92mg 투여군에서 2.5%, 위약 투여군에서 0.4%였다. 혈중 칼륨 감소는 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 0.4%, 15mg/92mg 투여군에서 1.0%, 위약 투여군에서 0.0%였다.
⓷ 혈청 크레아티닌
1년간의 위약‑대조임상시험에서 기저치 대비 복용량과 관련된 증가가 나타났으며, 4주에서 8주 사이에 정점에 도달했다가 감소하였으나 1년간의 치료 기간 동안 기저치 이상으로 상승 유지되었다. 치료기간 동안 0.3 mg/dL 이상의 혈청 크레아티닌 증가가 발생할 확률은 3.75mg/23mg 투여군에서 2.1%, 7.5mg/46mg 투여군에서 7.2%, 15mg/92mg 투여군에서 8.4%, 위약 투여군에서 2.0%였다. 기저치 대비 50% 이상의 혈청 크레아티닌이 증가한 비율은 3.75mg/23mg 투여군에서 0.8%, 7.5mg/46mg 투여군에서 2.0%, 15mg/92mg 투여군에서 2.8%, 위약 투여군에서 0.6% 였다.
신석증
1년간의 위약‑대조임상시험에서 신장결석증 발생률은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 1.2%, 위약 투여군에서 0.3% 였다.
이상반응에 따른 약물 중단
1년간의 위약‑대조 임상시험에서 3.75mg/23mg 투여군에서 11.6%, 7.5mg/46mg 투여군에서 11.6%, 15mg/92mg 투여군에서 17.4%, 위약 투여군에서 8.4%가 보고된 이상반응으로 인해 약물 치료를 중단했다. 치료중단을 야기한 가장 흔한 이상반응은 표 2에 나타난다.
표 2. 1% 이상에서 약물 치료 중단을 야기한 이상반응(1 year clinical trials)
2) 시판후조사
미국에서 이 약의 성분인 펜터민과 토피라메이트의 시판후 사용에 대하여 다음과 같은 이상반응이 보고되었다. 이러한 이상반응은 인구수를 특정하기 어려운 모집단으로부터 자발적으로 보고되기 때문에 발생 빈도를 정확하게 예측하거나 약물 노출과의 인과 관계를 확립하는 것이 항상 가능하지는 않다.
큐시미아
정신 장애 : 자살 생각, 자살 행동
안과 질환 : 급성폐쇄각녹내장, 안압 상승
펜터민
알레르기 이상반응 : 두드러기
심혈관 이상반응 : 혈압 상승, 허혈성 사건
중추 신경계 이상반응 : 다행감, 정신증, 진전
생식 이상반응 : 성욕의 변화, 발기부전
토피라메이트
피부질환 : 수포 피부 반응(다형 홍반, 스티븐존슨 증후군, 독성표피괴사용해 포함), 천포창
위장 장애 : 췌장염
간 질환 : 간 실조(사망자 포함), 간염
대사 장애 : 고암모니아혈증, 저체온증
안과 질환 : 황반증
5. 일반적 주의
1) 심박수 증가
이 약은 안정시 심박수를 증가시킬 수 있다.
이 약으로 치료한 과체중 및 비만 성인과 위약으로 치료한 과체중 및 비만 성인을 비교하였을 때, 이 약 치료군에서 기저치에 비해 분당 심박수가 5, 10, 15 및 20회 (bpm) 이상 증가한 환자의 비율이 높았다.
표 3은 최대 1년간의 임상연구에서 심박수의 상승을 보이는 환자의 수와 백분율을 나타낸다.
표 3. 단일 시점을 기준으로 기저치 심박수에서 심박수가 증가한 환자의 수와 백분율
지난 6개월 동안 심근경색이나 뇌졸중 병력이 있는 환자, 생명을 위협하는 부정맥 또는 울혈성심부전증이 있는 환자와 같은 심장 및 뇌혈관 질환 환자에 대해 특히 이 약의 치료로 인한 심박수 상승의 임상적 의미는 분명하지 않다.
이 약을 복용하는 모든 환자, 특히 심장 질환이나 뇌혈관 질환이 있는 환자 또는 이 약의 복용을 시작하거나 늘릴 때 안정시 심박수를 정기적으로 측정할 것을 권장한다. 이 약은 불안정한 심장 질환이나 뇌혈관 질환이 있는 환자를 대상으로 연구를 진행하지 않았으므로 이 환자들에 대한 사용은 권장하지 않는다.
환자는 이 약의 치료기간 동안 휴식을 취할 때 두근거림이나 고동치는 느낌을 경험하는 경우, 이 사실을 의료진에게 알려야 한다. 이 약을 복용하는 동안 안정시 심박수가 지속적으로 증가하는 환자의 경우, 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
2) 자살 행동과 자살 생각
이 약의 성분인 토피라메이트를 포함한 항경련제(AED)는 이러한 약물을 복용하는 목적과 상관없이 약물을 복용하는 환자에게 자살 충동이나 자살 행동 위험을 증가시킨다. 이 약으로 치료받는 환자는 우울증의 발생이나 증상의 악화, 자살 충동이나 자살 행동 및/또는 환자의 기분이나 행동의 비정상적인 변화에 대해 모니터링되어야 하며, 이러한 증상 또는 행동이 발현될 경우 즉시 의료전문가에게 보고될 수 있도록 한다. 자살 충동이나 자살 행동을 경험한 환자는 이 약의 복용을 중단한다.
자살 시도나 적극적인 자살 생각의 병력이 있는 환자는 이 약의 복용을 피해야 한다.
여러 적응증이 있는 11가지 항경련제에 대한 199개의 위약 대조 임상연구(단일요법 및 보조요법, 중간 치료 기간 12주)를 통합 분석한 결과, 항경련제 중 하나에 무작위로 배정된 환자는 자살 충동이나 행동이 위약에 무작위로 배정된 환자에 비해 약 2배의 위험도(보정 상대 위험도(adjusted relative risk) 1.8, 95% 신뢰 구간 [CI] 1.2, 2.7)를 나타냈다. 위약으로 치료한 환자 16,029명에서의 자살 행동이나 자살 생각의 예상 발생률은 0.24%인 것에 비해, 항경련제로 치료한 환자 27,863명에서 예상 발생률은 0.43%로, 치료받은 환자 530명당 약 1건의 자살 생각이나 행동의 증가가 나타났다. 임상시험에서 항경련제로 치료받은 환자에서는 4건의 자살이 있었고 위약으로 치료받은 환자에서는 한 건도 없었지만, 그 수가 너무 적어 자살과 관련된 항경련제 영향에 대해서는 결론을 내릴 수 없었다.
항경련제로 인한 자살 충동이나 행동 위험의 증가는 항경련제로 약물 치료를 시작한 후 빠르면 1주일 후에 관찰되었으며, 치료 기간 내내 지속되었다. 임상분석에 포함된 대부분의 임상시험들은 24주를 초과하지 않았으므로, 24주 이후의 자살 충동이나 행동의 위험을 평가할 수 없었다.
자살 충동이나 행동의 위험은 분석된 데이터에 사용된 약물에서 일반적으로 일치했다. 다양한 작용 기전과 다양한 범위의 적응증을 가진 항경련제에서 위험 증가가 확인되었다는 것은, 항경련제의 적응증과는 무관하게 모든 항경련제에 위험성이 있다는 것을 시사한다. 위험도는 분석된 임상시험에서 연령(5세에서 100세)에 따라 크게 변하지 않았다.
3) 급성 근시 및 2차성 폐쇄각녹내장
이 약의 성분인 토피라메이트로 치료받은 환자들에서 2차성 폐쇄각녹내장과 관련된 급성 근시 증후군이 보고되었으며, 그 증상에는 갑작스런 시력저하 및/또는 안통이 포함된다. 안과적인 검사시 근시, 좁아진 전방, 안 충혈 및 안압 상승이 관찰될 수 있다. 산동은 나타날 수도 있고 아닐 수도 있다. 이 증후군은 모양체상방 삼출 (supraciliary effusion)로 인한 수정체와 홍체의 전방 이동 및 2차성 폐쇄각녹내장과 관련이 있을 수 있다. 증상은 일반적으로 토피라메이트 치료 시작 후 1개월 이내에 발생하지만 치료 중 언제든지 발생할 수 있다. 증상에서 회복되기 위한 1차 치료는 이 약의 복용을 즉각 중단하는 것이다.
어떤 병에 의한 안압상승이라도 치료받지 않으면 영구 시력손실을 포함한 심각한 후유증을 가져올 수 있다.
이 약의 성분인 토피라메이트를 투여 받은 환자에게서 안압 상승과 독립적으로 시야결손이 보고되었다. 임상시험에서 이러한 반응의 대부분은 토피라메이트 중단 후 가역적이었다. 만일 이 약 치료 중 어느 때라도 시각 문제가 발생한다면 약물 중단을 고려해야 한다.
4) 기분 장애 및 수면 장애
이 약은 우울증과 불안, 불면증 등 기분 장애를 유발할 수 있다. 우울증 병력이 있는 환자는 이 약을 복용하는 동안 우울증 또는 기타 기분 장애의 재발 위험이 높아질 수 있다. 이 기분 장애와 수면 장애의 대부분은 자연적으로 치료되었거나 복용 중단 시 치료되었다. 임상적으로 유의미하거나 지속적인 증상이 나타나는 경우, 이 약의 복용량을 줄이거나 복용 중단을 고려해야 한다. 환자에게서 자살 생각이나 자살 행동 증상을 보이면 이 약의 복용을 중단해야 한다.
5) 인지 장애
이 약은 인지 기능 장애(예: 주의/집중력 장애, 기억곤란, 언어 장애, 특히 단어 선택의 어려움)을 유발할 수 있다.
이 약의 용량을 빠르게 적정하거나 초기에 고용량으로 복용하면 주의력, 기억력, 언어/단어 선택 어려움과 같은 인지기능 관련 이상반응 발생률을 높이는 경우가 있다.
이 약은 인지 기능을 손상시킬 가능성이 있기 때문에, 환자는 이 약의 치료가 악영향을 미치지 않는다고 확신할 수 있을 때까지 자동차를 포함한 위험한 기계를 조작할 때 주의하여야 한다. 인지 기능 장애가 지속되면 감량을 고려해야 하며, 중등도에서 중증까지의 증상, 불편함 또는 복용량 감소로 해결하지 못한 증상에 대해 이 약의 복용량 감소 또는 복용 중단을 고려해야 한다.
6) 대사성산증
이 약으로 치료한 환자에서 음이온 차의 변화 없는(non‑anion gap) 고염소혈증성 대사성 산증(만성 호흡성 알칼리혈증 없이 정상 범위 이하로 혈청 중탄산염 농도 저하)이 보고되었다. 신장질환, 심한 호흡기질환, 간질 중첩증, 설사, 수술, 케톤 식이 등 산증의 위험을 증가시킬 수 있는 상태 혹은 치료가 토피라메이트의 중탄산염 농도 저하 효과를 증가시킬 수 있다. 이 약과 탄산탈수효소억제제(예: 조니사미드, 아세타졸아미드 또는 디클로펜아미드)를 병용할 경우 대사성산증의 중증도를 증가시킬 수 있으며 또한 신장 결석 형성의 위험도도 높아질 수 있다. 따라서 대사성산증이 발생하기 쉬운 상태의 환자가 이 약을 다른 탄산탈수효소억제제와 병용하는 경우에는 대사성산증의 발생이나 악화 여부를 모니터링 하여야 한다.
급성 또는 만성 대사성산증의 일부 증상으로는 과다호흡, 피로 및 거식증과 같은 비특이적 증상, 또는 심장부정맥 또는 혼미를 포함한 보다 심각한 후유증이 있을 수 있다. 만성적이고 치료되지 않은 대사성산증은 신석증이나 신장석회증의 위험도를 높일 수 있으며, 골연화증(소아 환자에서 구루병이라고도 함) 및/또는 골절 위험이 높은 골다공증을 유발할 수 있다. 성장 및 뼈 관련 후유증에 대한 이 약의 효과는 장기적 위약 대조 임상 시험에서 체계적으로 조사되지 않았다.
이 약의 치료 전과 치료 중에 혈청 중탄산염을 포함한 전해질 측정이 권장된다. 이 약의 임상시험에서 혈청 중탄산염의 피크 감소는 4주째까지 발생했으며, 대부분 피험자에서는 시험약 변경 없이 56주째까지 중탄산염 교정이 있었다. 그러나 이 약의 복용 중 지속적인 대사산증이 발생하면 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
7) 크레아티닌 상승
이 약은 혈청 크레아티닌 증가를 유발할 수 있다. 4주에서 8주간의 치료 후에 혈청 크레아티닌의 피크 증가가 관찰되었다. 평균적으로 혈청 크레아티닌은 점차 감소했지만 기저치 크레아티닌 값보다 높게 유지되었다. 혈청 크레아티닌의 변화(측정된 GRF)는 치료를 중단하면 회복되지만, 만성적으로 치료시에 신장 기능에 미치는 영향에 대해서는 알려지지 않았다. 따라서 이 약 치료 시작 전과 치료 중에 혈청 크레아티닌 측정이 권장된다. 이 약의 복용 중에 크레아티닌의 지속적인 상승이 발생하면 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
8) 당뇨병 치료를 하는 제2형 당뇨 환자에서 저혈당증의 잠재적 위험
체중 감량은 인슐린 및/또는 인슐린 분비 촉진제(예: 설폰요소제)로 치료되는 제2형 당뇨 환자의 저혈당증 위험을 높일 수 있다. 이 약은 인슐린과의 병용 치료에 관해서는 연구되지 않았다. 제2형 당뇨 환자에 대해서는 이 약의 치료 시작 전과 치료 중에 혈당 수치를 측정할 것을 권장한다. 저혈당의 위험을 감소시키기 위해서 비포도당 의존성(non‑glucose‑dependent) 항당뇨병제의 약물 복용량 감소를 고려해야 한다. 이 약의 복용을 시작한 후 환자가 저혈당증을 일으키면 당뇨병치료제 처방을 적절히 변경해야 한다. 또한, 이 약 투여 시 당뇨병 환자에서 인슐린 요구량이 변경될 수 있으므로 당뇨병 환자에게 식이요법과 병행하여 이 약을 투여할 경우 인슐린 투여량을 조절하여야 한다.
9) 혈압강하제로 치료받는 환자에게서 저혈압의 잠재적 위험
혈압강하제로 치료를 받는 고혈압 환자의 경우, 체중 감량은 저혈압의 위험을 높일 수 있으며 어지러움, 어질어질함, 실신 등을 유발할 수 있다. 고혈압 치료를 받는 환자는 이 약의 치료 시작 전과 치료 중 혈압 측정을 권장한다. 이 약의 복용을 시작한 후 환자에게서 저혈압과 관련된 증상을 보이면 항고혈압제 처방을 적절하게 변경해야 한다.
10) 알코올을 포함한 중추신경 억제제로 인한 중추신경기능저하
펜터민 또는 토피라메이트와 알코올 또는 중추신경 억제제(예: 바르비탈계, 벤조디아제핀 및 수면제)를 병용하면 어지러움, 인지기능 관련 이상반응, 나른함, 어질어질함, 조정력 저하 및 졸림과 같은 중추신경기능저하나 이러한 약제의 기타 중추 매개 효과를 증가시킬 수 있다. 따라서 이 약은 알코올을 함께 복용하면 안 된다.
11) 갑작스런 복용 중단으로 인한 잠재적 발작
이 약의 성분인 토피라메이트의 갑작스런 복용 중단으로 인한 금단 현상은 발작이나 간질 병력이 없는 개인에게도 발작 증상을 일으키는 경우가 있다. 이 약의 즉각적인 복용 중단이 의학적으로 요구되는 상황에서는 환자에 대한 적절한 모니터링이 권장된다. 이 약 15mg/92mg 용량의 복용을 중단하는 환자는 발작을 일으킬 가능성을 줄이기 위해 권장하는 대로 그 복용량을 점차적으로 줄여야 한다.
12) 신장애 환자
이 약의 성분인 펜터민과 토피라메이트는 신장 배설을 통해 제거된다. 따라서 중등증(크레아티닌 청소율(CrCl)이 30 mL/min 이상 50 mL/min 미만) 또는 중증(CrCl 30 mL/min 미만) 신장애 환자에서 펜터민과 토피라메이트에 대한 노출이 더 높다. 두 환자군에서 모두 이 약의 용량을 조절한다.
이 약은 신장 투석 중에 있는 말기 신장질환 환자에 대해서는 연구되지 않았다. 이 환자군에서는 이 약의 사용을 피해야 한다.
13) 간장애 환자
경증(Child‑Pugh score 5 ‑ 6) 또는 중등증(Child‑Pugh score 7 ‑ 9) 간장애 환자에서 펜터민에 대한 노출은 건강한 지원자에 비해 더 높았다. 중등증의 간장애 환자에서는 이 약의 용량을 조정한다.
이 약은 중증 간장애 환자(Child‑Pugh score 10 ‑ 15)에 대해서는 연구되지 않았다. 이 환자군에서는 이 약의 사용을 피해야 한다.
14) 신장 결석
이 약의 복용은 신장결석 형성과 관련이 있다. 이 약의 성분인 토피라메이트는 탄산탈수효소활성(carbonic anhydrase activity) 을 억제하고 뇨중 구연산의 배설을 감소시켜, 뇨의 pH를 높임으로써 신장 결석 형성을 촉진한다.
탄산탈수효소(예: 조니사미드, 아세타졸아미드, 메타졸아미드) 를 억제하는 다른 약물과 함께 이 약을 사용하지 않는다.
케톤식이요법(Ketogenic Diet)을 하는 환자의 토피라메이트 사용은 또한 신장 결석 형성의 가능성을 증가시키는 생리적 환경을 초래할 수 있다.
수분 섭취량을 늘려서 소변량을 늘리면 신장 결석 형성에 관련있는 물질의 농도를 낮출 수 있다. 운동이나 높은 기온에 노출되기 전 및 중에 적절한 수분 공급을 하는 것은 열과 관련된 이상반응의 위험을 줄일 수 있다
15) 땀분비 감소증 및 고체온증
이 약의 성분인 토피라메이트의 복용으로 입원이 필요할 수도 있는 땀분비 감소증이 보고되었다. 땀분비 감소와 정상보다 높은 체온 증가가 특징으로 나타난다. 일부 사례는 온도가 상승한 환경에 노출된 이후에 토피라메이트 복용과 함께 보고되었다.
이 약으로 치료받는 환자는 신체 활동 중, 특히 더운 날씨에 땀분비 감소와 체온 증가를 모니터링 해야 한다. 이 약을 열 관련 질환에 걸리기 쉬운 환자에게 다른 약제와 함께 처방할 때는 각별히 주의해야 한다. 이들 약제에는 다른 탄산탈수효소억제제 및 항콜린성 작용 약제가 포함되나 이에 한정되지는 않는다.
16) 저칼륨혈증
이 약은 탄산탈수효소활성의 억제를 통해 저칼륨혈증의 위험을 높일 수 있다. 또한 이 약을 푸로세미드(루프이뇨제) 또는 히드로클로로티아지드 (티아지드 유사 이뇨제)와 같은 비‑칼륨보존이뇨제(non‑potassium sparing diuretics)와 병용시 칼륨 손실을 더욱 증가시킬 수 있다. 이 약 처방 시 환자는 저칼륨혈증에 대해 모니터링되어야 한다.
17) 모니터링: 실험실적 검사
이 약은 무작위, 이중 맹검, 위약 대조 임상연구에서 여러 실험실적 분석 결과의 변화를 일으키는 경우가 있었다.
기저시점 및 치료 기간 중 주기적으로 중탄산염, 크레아티닌, 칼륨 및 혈당을 포함하는 혈액 화학 프로필을 확보해야 한다.
18) 남용
이 약의 주성분인 펜터민은 약물 남용의 가능성이 있다. 펜터민은 암페타민과 화학적 및 약리학적으로 관련이 있다. 암페타민과 기타 각성제는 광범위하게 남용되어 왔으며, 이 약을 체중 감량 프로그램의 일부로 포함시키는 것이 바람직하다고 평가할 때 펜터민의 남용 가능성을 염두에 두어야 한다. 암페타민 및 관련 약물(예: 펜터민)의 남용은 약물 사용 조절 장애와 심각한 사회적 기능 장애를 일으키는 경우가 있다. 이 약의 복용량을 권장 용량 보다 여러 번 늘린 환자에 대한 보고가 있다.
19) 의존성
이 약은 육체적 약물 의존 가능성에 대해 체계적으로 연구되지는 않았다. 육체적 약물 의존성은 반복되는 약물 복용에 대해서 생리적으로 적응하려는 상태이다. 육체적 약물 의존성은 약물 사용의 갑작스런 중단 또는 약물의 현저한 (복)용량 감소 이후에 약물 종류별 금단 현상으로 나타난다.
이 약의 각 주성분에 대한 육체적 약물 의존 가능성에 대한 정보는 한정되어 있다. 토피라메이트의 경우, 갑작스러운 약물 중단은 발작이나 간질 병력이 없는 환자에서도 발작이 일어나는 경우가 있었다. 펜터민의 경우, 장기적인 고용량 복용 후 갑작스런 중단은 극심한 피로와 정신적 우울증을 야기한다. 수면의 뇌파에도 변화가 나타난다. 따라서 이 약의 신속한 복용 중단이 필요한 상황에서는 적절한 의학적 모니터링이 권장된다.
20) 이 약은 최근 1년 이내에 다른 식욕억제제를 사용한 환자에게는 투여가 권장되지 않으므로 주의한다.
21) 토피라메이트를 투여 받은 환자에서 뇌병증을 동반 혹은 동반하지 않은 고암모니아혈증이 보고되었다. 고암모니아혈증은 토피라메이트와 발프로산을 병용 투여한 경우 더 빈번하게 보고되었다(상호작용 항 참조). 고암모니아혈증은 임상적으로 증상이 없을 수 있으나, 고암모니아혈증성 뇌병증은 기면 또는 구토를 동반한 의식수준 및/또는 인지기능에 있어서 급격한 변화를 자주 보인다. 이 약과 관련하여 설명할 수 없는 기면, 구토 혹은 정신상태의 변화가 발생한 환자의 경우, 고암모니아혈증성 뇌병증을 고려하여 혈중 암모니아 수치를 측정하는 것이 권장된다.
6. 상호작용
1) 모노아민산화효소억제제
고혈압 발생의 위험성 때문에 모노아민 산화효소 억제제(monoamine oxidase inhibitors) 투여기간 동안 또는 투여 후 14일 동안 펜터민 복용을 금한다.
2) 경구피임약
건강한 지원자와는 달리 비만환자에게서 에티닐에스트라디올(에스트로겐 성분) 35 ug과 노르에틴드론 (프로게스틴 성분) 1 mg을 함유한 경구 피임약의 단회 용량과 이 약 15 mg/92 mg의 1일 1회 병용 투여는 에티닐에스트라디올의 노출을 16% 감소시키고, 노르에틴드론의 노출을 22% 증가시켰다.
이 연구가 피임 효과에 대한 상호 작용의 영향을 구체적으로 다루지는 않았지만, 임신의 위험도가 증가할 것이라고 예상되지는 않는다. 피임 효과의 주된 결정 요인은 복합 경구 피임약의 프로게스틴 성분이므로 높아진 프로게스틴 노출이 유해할 것으로 예상되지는 않는다.
그러나 불규칙한 출혈(점상출혈)은 자궁 내막을 안정화시키는 프로게스틴에 대한 노출 증가와 에스트로겐 노출 감소로 인해 더 자주 발생할 수 있다. 환자는 출혈이 발견되면 복합 경구 피임약을 중단하지 말아야 한다는 사실을 통보 받아야 한다. 그러나 점상출혈이 불편할 경우 의사에게 알려야 한다.
3) 알코올을 포함한 중추신경억제제
이 약과 알코올 또는 기타 중추신경억제제과의 특정 약물상호작용에 관한 연구는 수행되지 않았다. 펜터민 또는 토피라메이트와 알코올 또는 중추신경억제제(예: 바르비탈류, 벤조디아제핀 및 수면제)의 병용 투여는 어지러움 또는 인지 이상반응 또는 이러한 약물의 중추 매개 효과와 같은 중추신경기능저하를 증가시킬 수 있다. 따라서 이 약을 알코올이나 다른 중추신경억제제와 함께 사용하는 경우 환자는 중추신경기능저하나 이상반응 위험이 증가할 가능성에 대해 상담받아야 한다.
4) 비‑칼륨보존이뇨제
비‑칼륨보존이뇨제와 함께 이 약을 병용투여하면 이러한 이뇨제의 칼륨 손실 작용이 증가할 수 있다. 토피라메이트 단일제와 히드로클로로티아지드 단일제를 병용하는 경우 토피라메이트의 Cmax 및 AUC가 각각 27% 및 29% 증가하는 것으로 나타났다. 비‑칼륨보존 의약품과 이 약을 병용 처방할 때, 환자의 저칼륨혈증에 대해 모니터링해야 한다.
5) 항경련제
간질 환자가 토피라메이트와 페니토인 또는 카르바마제핀을 병용하는 경우, 토피라메이트를 단독 복용하는 것에 비해 토피라메이트의 혈장 농도가 각각 48%와 40% 감소했다.
발프로산과 토피라메이트의 병용투여는 뇌병증과는 무관하게 고암모니아혈증을 일으키는 경우가 있다. 또한 발프로산과 토피라메이트를 병용한 환자에서 고암모니아혈증과 무관하게 저체온증이 나타나는 경우가 있다. 저체온증이나 뇌병증의 발병이 보고되는 경우, 환자의 혈중 암모니아 수치를 검사하는 것이 권장된다.
6) 탄산탈수효소억제제
이 약의 성분인 토피라메이트를 다른 탄산탈수효소억제제(예: 조니사미드, 아세타졸아미드,디클로펜아미드)와 함께 사용하면 대사성산증의 중증도가 증가할 수 있으며 신장 결석 형성의 위험도 증가할 수 있다. 이 약을 탄산탈수효소를 억제하는 다른 약물과 함께 사용해서는 안된다.
7) 피오글리타존
이 약의 성분인 토피라메이트와 피오글리타존의 병용투여 임상시험에서 피오글리타존과 활성대사산물 노출의 손실이 보고되었다. 이 결과의 임상적유의성은 알려지지 않았다. 그러나 피오글리타존 치료 중 이 약의 병용 또는 이 약 치료 중 피오글리타존 병용 시, 당뇨병 환자의 상태를 적절하게 통제하기 위해 환자에 대한 일상적인 모니터링 시 세심한 주의가 필요하다.
8) 아미트리프틸린
일부 환자는 이 약의 성분인 토피라메이트 복용 시 아미트리프틸린 농도가 크게 증가할 수 있다. 이 약과 병용투여 시 아미트리프틸린 용량 조절은 혈청 수치가 아닌 환자의 임상 반응에 따라 이루어져야 한다.
7. 임부 및 수유부에 대한 투여
1) 임부
이 약은 임부에게 사용해서는 안된다. 이 약은 태아에게 유해한 영향을 줄 수 있으며, 이 약 사용으로 인한 체중 감량은 임신한 여성에게 잠재적인 이득이 되지 않는다. 역학자료에서는 이 약의 성분인 토피라메이트에 임신 초기 3개월 동안 노출시 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다. 여러 종의 임신한 동물이 임상적으로 적절한 용량의 토피라메이트를 복용했을 때 두개 및 안면 결손을 포함한 구조적 기형 및 자궁 내 태아 체중 감소가 후손에서 발생했다.
환자가 임신 중에 이 약물을 복용하거나 이 약물 복용 중에 임신한 경우, 치료를 즉시 중단하고 태아에게 미칠 잠재적인 위험이 있음을 환자에게 알려야 한다.
임상적 고찰
구순구개 파열은 임신 5주부터 9주까지 발생한다. 입술은 임신 5주에서 7주 사이에 형성되며, 구개(입천장)는 임신 6주부터 9주 사이에 형성된다.
임신 중 모체 조직에서 발생하는 필수적인 체중 증가 때문에 이미 과체중이거나 비만인 임산부 뿐 아니라, 모든 임산부의 경우 최소한의 체중 증가 및 체중 감량이 되지 않는 것이 권장된다.
이 약은 대사성산증을 일으킬 수 있다. 토피라메이트 유발 대사성산증의 영향은 임신에 대해서는 연구되지 않았다. 그러나 다른 원인으로 인한 임신 중 대사성산증은 태아의 성장 감소, 태아에게 공급되는 산소 감소 및 태아 사망을 유발할 수 있으며, 분만을 견딜 수 있는 태아의 능력에 영향을 미칠 수 있다.
임상 데이터
임신 중에 이 약의 구성성분인 토피라메이트 노출로 인한 주요 선천성 기형 및 구순구개 파열 위험을 평가한 데이터는 North American Anti‑Epileptic Drug (NAAED) Pregnancy Registry 및 몇 가지 대규모 후향적 역학 연구에서 확인할 수 있다. NAAED Pregnancy Registry는 구순구개 파열 위험이 9.60(95% CI 3.60 ‑ 25.70)으로 증가할 것으로 예상했다. 대규모 후향적 역학 연구에서 임신 중 토피라메이트 단일요법 노출이 구순구개 파열 위험성의 약 2~5배 증가와 연관이 있었다. (표 4). FORTRESS 연구에서는 임신 초기에 토피라메이트에 노출된 1,000명의 유아 당 구순구개 파열 증상의 위험성이 1.5배 (95% CI = ‑1.1 ~ 4.1) 증가가 관찰되었다.
표 4. 자궁 노출 및 구순구개 파열 및 주요 선천성 기형에서의 토피라메이트 연관성 평가 연구 요약
동물 데이터
펜터민/토피라메이트
태아 발육 연구는 랫드와 토끼에서 펜터민과 토피라메이트 병용 요법으로 수행되었다. 기관이 형성되는 기간에 랫드에게 투여된 펜터민과 토피라메이트는 태아의 체중 감소를 가져 왔지만, 최고용량인 펜터민 3.75 mg/kg과 토피라메이트 25 mg/kg에서 태아 기형을 일으키지 않았다. (각 주성분에 대해 AUC 예상치에 근거한 인체최대권장용량의 약 2배). 토끼를 대상으로 한 유사한 연구에서는 AUC에 근거한 인체최대권장용량의 약 0.1배(펜터민) 및 1배(토피라메이트)의 임상 노출이 있었을 때, 태아 발달에 대한 어떠한 영향도 관찰되지 않았다. 랫드와 토끼에서 이 용량으로 유의미하게 낮은 모체의 체중 증가가 기록되었다.
산전/산후 발달 연구는 펜터민과 토피라메이트 치료의 병용 요법으로 랫드에서 시행되었다. 1.5 mg/kg/day의 펜터민과 10 mg/kg/day의 토피라메이트 (AUC 기준 인체최대권장용량에서 각각 약 2배와 3배의 임상 노출)로 기관형성에서 수유기까지 치료한 랫드의 모체 또는 자손에 대한 유해한 영향은 없었다. 고용량 11.25 mg/kg/day의 펜터민과 75 mg/kg/day의 토피라메이트(AUC 기준 최대 임상 용량의 약 5배와 6배)를 투여하면 모체 체중 증가의 감소와 자손 독성을 초래한다. 자손에 대한 영향으로는 출생 후 자손 생존률 감소, 사지 및 꼬리의 기형 증가, 자손 체중 감소 및 학습, 기억 또는 수정과 생식에는 영향을 미치지 않으며 성장, 발달 및 성적 성숙을 지연시켰다. 사지 및 꼬리 기형은 토피라메이트 단독으로 수행한 동물 연구의 결과와 일치했다.
펜터민
펜터민에 대한 동물 생식 연구는 수행되지 않았다. 펜터민/토피라메이트 병용 요법으로 실시한 랫드 대상 연구로 부터 제한된 데이터는 펜터민 단독으로는 최기형성을 보이지 않았지만 AUC 기준으로 이 약의 인체최대권장용량의 5배에서 저체중과 태아의 생존률 감소를 보였다.
토피라메이트
토피라메이트는 임상적으로 적절한 용량에서 최기형성을 포함한 발달 독성을 유발한다.
2) 분만
이 약이 인간의 분만에 미치는 영향은 알려지지 않았다. 산모 및/또는 태아에서 이 약으로 유도되는 대사성산증의 발달은 분만을 견딜 수 있는 태아의 능력에 영향을 미칠 수 있다.
3) 수유부
토피라메이트와 암페타민(펜터민은 암페타민과 유사한 약리학적 활성 및 화학적 구조를 가짐)이 모유에서 배출되기 때문에 이 약이 사람의 모유에 존재할 수 있다. 신생아의 모유 수유에 대한 심각한 이상반응의 잠재적 위험성때문에, 산모에 대한 약물 처방의 중요성을 고려하여 수유를 중단할 것인지 약물 투여를 중단할 것인지에 대한 여부를 결정해야 한다.
4) 가임여성
이 약은 태아에 유해할 수 있다. 임신 등록 및 역학 연구의 자료에 따르면 임신 초기에 이 약 성분인 토피라메이트에 노출된 태아는 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다. 이 약 치료 중 임신한 여성은 즉시 치료를 중단하고 의료진에게 알려야 한다.
임신 테스트
가임기 여성은 이 약의 치료를 시작하기 전에 임신 테스트의 결과가 음성임을 확인해야 하고, 이 약으로 치료하는 동안에는 매달 한 번씩 임신 테스트를 해야 한다.
피임
이 약 치료 중 가임 여성은 효과적인 피임법을 사용해야 한다.
8. 소아에 대한 투여
18세 미만 소아 환자를 대상으로 한 이 약의 안전성과 효과는 입증되지 않았으며, 이 약의 사용은 소아 환자에게 권장되지 않는다. 이 약의 성분인 토피라메이트를 복용한 소아 환자에서 나타나는 심각한 이상반응으로는 급성 녹내장, 땀분비 감소증 및 고체온증, 대사성산증, 인지 및 신경 정신적 반응, 고암모니아혈증 및 뇌병증, 신결석이 있다.
어린 동물 연구
이 약에 대한 어린 동물 연구는 수행되지 않았다. 어린 연령(출생 후 12 ~ 50일)의 랫드에게 토피라메이트(30, 90 또는 300 mg/kg/day)를 경구 투여했을 때, 가장 높은 용량에서 수컷 랫드의 뼈 성장판 두께가 감소했다.
9. 고령자에 대한 투여
이 약의 임상 시험에서 65세 이상 환자는 총 254명(7%)이었다. 고령 피험자와 젊은 연령의 피험자 사이에 안전성이나 효과에 전반적인 차이는 없었지만, 일부 고령자에게서 나타나는 더 큰 과민반응은 배제할 수 없었다.
이 약의 임상 연구에는 젊은 연령의 피험자와 다르게 반응하는지를 판단할 수 있을 정도의 충분한 65세 이상 피험자 수가 포함되지 않았다. 일반적으로 고령 환자에 대한 투여량 선택은 신중해야 하며, 투여량 선택은 보통 투여 범위의 최하위부터 시작하며, 간, 신장 또는 심장 기능 저하와 동반 질환 또는 기타 약물 치료의 빈도가 높아지는 것을 반영해야 한다.
10. 신장애 환자
건강한 지원자와 비교할 때 C‑G 공식(Cockcroft‑Gault equation)에 의해 평가된 중등증 및 중증 신장애 환자에서 펜터민과 토피라메이트에 대해 더 높은 노출을 보였다.
경증 신장애가 있는 환자에게는 용량 조절이 필요하지 않다. 중등증(30 이상 50 mL/min 미만의 크레아티닌 청소율[CrCl]) 및 중증 (30 mL/min 미만의 CrCl) 신장애 환자에게 있어 복용량은 1일 1회 7.5 mg/46 mg을 초과해서는 안 된다.
이 약은 신장 투석 중에 있는 말기 신장질환 환자에 대해서는 연구되지 않았다. 이러한 환자군에서는 이 약의 사용을 피해야 한다.
11. 간장애 환자
경증(Child‑Pugh 5 ‑ 6) 및 중등증(Child‑Pugh 7 ‑ 9) 간장애 환자에서 펜터민에 대한 노출은 건강한 지원자에 비해 더 높았다. 이 약 성분인 토피라메이트에 대한 노출은 경증 및 중등증의 간장애 환자와 건강한 지원자 간에 유사했다.
경증의 간장애가 있는 환자에게는 용량 조절이 필요하지 않다. 중등증의 간장애가 있는 환자의 경우 1일 1회 7.5 mg/46 mg을 초과해서는 안 된다.
이 약은 중증 간장애 환자(Child‑Pugh score 10 ‑ 15)에 대해서는 연구되지 않았다. 이러한 환자군에서는 이 약의 사용을 피해야 한다.
12. 과량투여시의 처치
이 약을 과다 복용하는 경우, 과다 복용한지 얼마 되지 않았다면 즉시 위세척이나 구토를 통해 위를 비워야 한다. 환자의 임상 징후와 증상에 따라 적절한 보조요법(supportive treatment)이 제공되어야 한다.
급성 펜터민 과다 복용은 불안, 진전, 과다반사, 빠른 호흡, 착란, 공격성, 환각 그리고 공황 상태를 일으키는 경우가 있다. 피로와 우울증은 대개 중추 자극(central stimulation)에 의한다. 심혈관 영향에는 부정맥, 고혈압 또는 저혈압, 순환허탈 (circulatory collapse) 등이 있다. 위장 증상으로는 오심, 구토, 설사, 복부경련이 있다. 치명적인 중독은 보통 경련과 혼수상태로 이어진다. 식욕억제를 동반하는 만성 중독 증상은 심각한 피부병, 현저한 불면증, 과민성, 과행동증 및 성격 변화를 포함한다. 만성 중독의 심각한 징후는 정신증이며, 종종 임상적으로 정신분열증과 구별할 수 없다.
급성 펜터민 중독의 관리는 주로 증상을 관찰하는 것이며 세척과 바르비탈계 약물을 통한 진정이 포함된다. 뇨의 산성화는 펜터민 배설을 증가시킨다. 이 약의 과량 투여로 인한 급격하고 심각한 고혈압에는 가능한 신속히 펜톨아민 정맥주사하는 것이 추천된다.
토피라메이트 과다 복용으로 심각한 대사성산증이 초래되었다. 다른 징후와 증상으로는 경련, 졸림, 언어 장애, 시야흐림, 복시, 정신기능 저하, 졸음증, 협조이상, 혼미, 저혈압, 복통, 초조, 어지러움, 우울증이 있다. 대부분의 경우 임상 결과는 심각하지 않았지만, 다량의 토피라메이트를 포함한 여러 종의 약물 과다 복용 후 사망이 보고되었다. 96 ~ 110 그램의 토피라메이트를 복용한 환자는 20 ~ 24시간 지속되는 혼수상태로 입원한 다음 3 ~ 4일 후에 완전히 회복되었다.
약용탄은 시험관내에서 토피라메이트를 흡착하는 것으로 나타났다. 혈액 투석은 체내에서 토피라메이트를 제거하는 효과적인 수단이다.
13. 보관 및 취급상의 주의사항
1) 어린이의 손이 닿지 않는 곳에 보관하도록 주의한다.
2) 다른 용기에 바꾸어 넣는 것은 사고원인이 되거나 품질 유지 면에서 바람직하지 않으므로 주의한다.
14. 전문가를 위한 정보
1) 약력학적 정보
암페타민의 전형적인 작용에는 중추 신경계 자극 및 혈압 상승이 포함된다. 타키필락시스(급성 내성)와 내성은 이러한 현상이 발견된 이 부류의 모든 약물에서 입증되었다.
심장 전기생리학
무작위, 이중 맹검, 위약 대조 및 활성 대조군(목시플록사신 400 mg) 및 평행군/교차, QT/QTc 연구를 통해 QTc 간격에 대한 이 약의 영향을 평가했다. 총 54명의 건강한 피험자에게 정상 상태에서 이 약 7.5 mg/46 mg을 투여한 다음 정상 상태에서 이 약 22.5 mg/138 mg으로 적정했다. 이 약 22.5 mg/138 mg [이 약 7.5 mg/46 mg의 펜터민과 토피라메이트 최대 농도(Cmax)에 대해 각각 4배 및 3배 높은 최대치료용량] 는 QTc의 기저치로부터의 변화를 측정했을 때 심장 재분극에 영향을 미치지 않았다.
사구체여과율(GFR)
건강한 비만 남성 및 여성은 이 약(1~3일 3.75mg/23mg, 4~6일 7.5mg/46mg, 7~9일 11.25mg/69mg, 10~28일 15mg/92mg)을 4주간 복용했다. 피험자의 사구체여과율은 이오헥솔 청소율을 통해 평가되었다. 평균적으로 이 약 치료기간동안 사구체여과율은 감소하였고 이 약 복용 중단 후 4주 이내에 기저치로 회복되었다.
<BR><SPAN style="FONT‑SIZE: 11pt">2) 약물동력학적 정보</SPAN>
펜터민
이 약 단회 용량 15 mg/92mg을 경구 투여한 결과, 평균 혈장 펜터민 최대 농도(Cmax), Cmax 까지의 시간(Tmax), 투약시간부터 최종 혈중농도 정량시간 t까지의 혈중농도‑시간곡선하면적(AUC0‑t), 투약시간부터 무한시간까지의 혈중농도‑시간곡선하면적 (AUC0‑∞)은 각각 49.1ng/mL, 6시간, 1990ng▪hr/mL 및 2000ng▪hr/mL이다. 이 약 15mg/92mg에 대한 고지방 식이는 펜터민 약물동력학에 영향을 미치지 않는다. 펜터민 약물동력학은 대략 이 약 3.75mg/23mg에서 펜터민 15mg/토피라메이트 100mg까지 용량에 비례한다. 펜터민/토피라메이트 15/100mg 복합제 캡슐을 정상 상태에서 투여할 때, AUC 및 Cmax의 평균 펜터민 축적 비는 모두 약 2.5이다.
토피라메이트
이 약 단회 용량 15mg/92mg을 경구 투여한 결과, 평균 혈장 토피라메이트의 Cmax 및 Tmax, AUC0‑t 및 AUC0‑∞는 각각 1020 ng/mL, 9시간, 61600 ng▪hr/mL 및 68000 ng▪hr/mL이었다. 이 약 15mg/92mg에 대한 고지방 식이는 토피라메이트 약물동력학에 영향을 미치지 않는다. 토피라메이트 약물동력학은 대략 이 약 3.75mg/23mg에서 펜터민 15mg/토피라메이트 100mg까지 용량에 비례한다. 펜터민 15mg/토피라메이트 100mg 투여량의 복합제 캡슐을 정상 상태에서 투여할 때, AUC 및 Cmax의 평균 토피라메이트 축적 비는 모두 약 4.0이다.
분포
펜터민
펜터민은 17.5%의 혈장 단백질과 결합한다. 예상되는 펜터민 분포 용적(Vd/F)은 개체군 약물동력학적 분석 결과 348 L이다.
토피라메이트
토피라메이트는 0.5~250μg/mL 혈중농도범위에서 15‑41% 혈장 단백질과 결합한다. 혈중 토피라메이트가 증가함에 따라 결합 분획(fraction bound)이 감소했다. 개체군 약물동력학적 분석을 통해 추정된 토피라메이트 Vc/F(중앙 구획의 용적) 및 Vp/F(말초 구획의 용적)는 각각 50.8 L 및 13.1 L이다.
신진 대사 및 배설
펜터민
펜터민은 두 가지 대사경로, 즉 방향족 고리(aromatic ring)에 p‑hydroxylation과 지방족 측쇄(aliphatic side chain)에 N‑oxidation을 가지고 있다. Cytochrome P450(CYP) 3A4는 주로 펜터민을 대사하지만 광범위한 신진 대사를 보이지는 않는다. Monoamine oxidase(MAO)‑A와 MAO‑B는 펜터민을 대사하지 않는다. 단독 투여 시 뇨에서 대사되지 않은 70 ~ 80%의 펜터민이 존재한다. 평균 펜터민의 말단 반감기는 약 20시간이다. 개체군 약물동력학적 분석을 통해 예상되는 펜터민 경구 청소율(CL/F)는 8.79L/h이다.
토피라메이트
토피라메이트는 광범위한 신진 대사를 보이지 않는다. 6개의 토피라메이트 대사물질(하이드록실화(hydroxylation), 가수분해(hydrolysis) 및 글루크론산화 (glucuronidation)을 통한)이 존재하며, 그 중 어떤 것도 투여된 용량의 5% 이상을 차지하지 않는다. 단독투여시 뇨에서 대사되지 않은 70% 용량의 토피라메이트가 존재한다. 평균 토피라메이트의 말단 반감기는 약 65시간이다. 개체군 약물동력학적 분석을 통해 예상되는 토피라메이트 CL/F는 1.17L/h이다.
특정환자집단
신장애
정상적인 신장 기능을 가진 건강한 지원자와 비교하여 만성 신장애 정도가 다양한 환자에서 이 약 15 mg/92 mg의 약물동력학을 평가하기 위해 단회투여, 공개 임상시험이 수행되었다. 이 연구에는 크레아티닌 청소율을 기준으로 경증(50 이상 80 mL/min 미만), 중등증(30 이상 50 mL/min 미만) 및 중증(30 mL/min 미만)으로 분류된 신장애 환자가 포함되어 있다. 크레아티닌 청소율은 C‑G 공식(Cockcroft‑Gault equation)에 기초하여 혈청 크레아티닌으로부터 추정하였다.
건강한 지원자와 비교했을 때, 펜터민의 AUC0‑inf는 중증, 중등증 및 경증의 신장애 환자에서 각각 91%, 45%, 22%였다. 펜터민 Cmax는 2 ~ 15% 더 높았다. 건강한 지원자와 비교했을 때, 토피라메이트 AUC0‑inf는 중증, 중등증 및 경증의 신장애 환자에서 각각 126%, 85%, 25%였다. 토피라메이트 Cmax는 6 ~ 17% 더 높았다. 펜터민 또는 토피라메이트 Cmax 또는 AUC와 크레아티닌 청소율 사이의 반비례 관계가 관찰되었다. 이 약은 신장 투석 중인 말기 신장질환 환자에서는 연구되지 않았다.
간장애
경증(Child‑Pugh score 5 ‑ 6)와 중등증(Child‑Pugh score 7 ‑ 9)의 간장애 환자에 비해 정상 간 기능을 가진 건강한 지원자에서의 이 약 15 mg/92 mg의 약물동력학을 평가하기 위해 단회투여, 공개 임상시험이 수행되었다. 경증 및 중등증의 간장애 환자에서, 펜터민 AUC는 건강한 지원자와 비교하여 37% 및 60% 더 높았다. 토피라메이트의 약물동력학은 건강한 지원자와 비교할 때 경증 및 중등증의 간장애 환자에서는 영향을 받지 않는다. 이 약은 중증 간장애 환자(Child‑Pugh score 10‑15)에서는 연구되지 않았다.
약물상호작용
약물상호작용의 시험관내 평가
펜터민
펜터민은 CYP 동종 효소 CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 및 CYP3A4의 억제제가 아니며 모노아민 산화 효소의 억제제도 아니다. 펜터민은 CYP1A2, CYP2B6 및 CYP3A4의 유도 인자가 아니다. 펜터민은 P‑당단백질(P‑glycoprotein) 기질이 아니다.
토피라메이트
토피라메이트는 CYP 동종 효소 CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2D6, CYP2E1 및 CYP3A4/5의 억제제가 아니다. 그러나, 토피라메이트는 CYP2C19의 경도(mild) 억제제이다. 토피라메이트는 CYP3A4의 경도(mild) 유도 인자이다. 토피라메이트는 P‑당단백질(P‑glycoprotein) 기질이 아니다.
다른 약물에 미치는 펜터민/토피라메이트의 영향
표 6. 펜터민/토피라메이트가 병용투여 약물의 약물동력학에 미치는 영향
표 7. 병용투여 약물이 펜터민/토피라메이트의 약물동력학에 미치는 영향
항경련제
간질 환자를 대상으로 한 대조 임상 약물동력학 연구에서 토피라메이트와 표준 항경련제(AED) 간의 잠재적인 상호 작용을 평가했다. 이러한 상호 작용이 평균 혈장 AUC에 미치는 영향을 표 7에 요약했다.
표 8에서 두 번째 열(AED 농도)은 토피라메이트가 추가될 때 첫 번째 열에 나열된 항경련제(AED) 농도에 어떤 현상이 발생하는지를 설명한다. 세 번째 열(토피라메이트 농도)은 토피라메이트 단독 투여 시 실험 환경에서의 토피라메이트 농도를 첫 번째 열에 기재된 약물의 병용투여가 어떻게 변화시키는지를 설명한다.
표 8. 토피라메이트와의 항경련제(AED) 상호 작용 요약
단회 투여 연구에서 토피라메이트의 병용투여로 혈장농도곡선 아래 혈청 디곡신 면적(AUC)이 12% 감소되었다. 이러한 관찰결과에 대한 임상적 관련성은 확립되지 않았다.
히드로클로로티아지드(HCTZ)
건강한 지원자를 대상으로 실시한 약물 상호작용 연구는 정상 상태에서의 히드로클로로티아지드(25 mg 매24시간마다) 및 토피라메이트(96 mg 매12시간마다) 단독 및 병용 투여 후 약물동력학을 평가하였다.
이 연구 결과 HCTZ을 추가하였을 때 토피라메이트의 Cmax가 27%, AUC가 29% 증가되었다. 이러한 변화의 임상적 유의성은 알려져 있지 않다. 정상 상태에서의 HCTZ의 약물동력학은 토피라메이트의 병용으로 유의한 영향을 받지 않았다. 토피라메이트와 HCTZ 투여 후 혈청 칼륨의 저하가 관찰되었으며 두 약물을 병용 투여했을 때 더 크게 나타났다.
피오글리타존
건강한 자원자를 대상으로 한 약물상호작용 연구에서 7일 동안 토피라메이트(96mg 1일 2회)와 피오글리타존(30mg 1일)을 단독 및 병용 투여 후 정상상태에서의 약물동력학을 평가하였다. 피오글리타존의 AUCt,ss가 15% 저하되었으며 Cmax,ss에는 변화가 관찰되지 않았다. 이 결과는 통계적으로 유의하지 않았다. 활성 수산화‑대사체의 Cmax,ss와 AUCt,ss는 각각 13%, 16% 저하되었으며, 활성 keto‑대사체의 Cmax,ss와 AUCt,ss는 60% 저하되었다. 이 결과의 임상적 유의성은 알려져 있지 않다. 피오글리타존 치료를 받는 환자에 이 약을 투여하거나 이 약을 투여받는 환자에 피오글리타존을 투여할 때 당뇨가 적절히 조절되고 있는지 모니터하는데 주의한다.
글리부리드
제2형 당뇨병 환자를 대상으로 한 약물상호작용 연구에서 글리부리드 단독(5mg/day) 및 토피라메이트와 병용(150mg/day)시의 정상상태 약물동력학을 평가하였다. 토피라메이트 투여 중 Cmax는 22% 감소하였고 글리부리드 AUC24가 25% 감소하였다. 또한, 활성 대사체 4‑trans‑히드록시글리부리드(M1) 및 3‑cis‑히드록시글리부리드(M2)의 전신노출은 각각 13% 및 15%까지 저하되었고, Cmax는 각각 18%와 25% 감소했다. 토피라메이트의 정상상태 약물동력학은 글리부리드의 병용투여에 영향을 받지 않았다.
리튬
토피라메이트 200 mg/day 투여 중 환자에서 리튬의 약물동력학은 영향을 받지 않았다. 그러나 토피라메이트를 600 mg/day까지 투여한 후 리튬의 전신 노출이 증가하는 것으로 나타났다(Cmax는 27%, AUC는 26%). 고농도의 토피라메이트와 병용 투여시 리튬 수치를 모니터링해야 한다.
할로페리돌
할로페리돌(5mg) 단회 투여의 약물동력학은 13명의 건강한 성인(남성 6명, 여성 7명)에서 토피라메이트(12시간마다 100mg)의 반복 투여에 의한 영향을 받지 않았다.
아미트립틸린
토피라메이트 200 mg/day를 투여받은 일반인 18명(남성 9명, 여성 9명)에서 아미트립틸린(하루 25mg)의 AUC와 Cmax가 12% 증가했다. 일부 피험자에서 토피라메이트를 복용 시 아미트립틸린 농도가 크게 증가할 수 있으며, 아미트립틸린 용량 조절은 혈청 수치가 아닌 환자의 임상 반응에 따라 이루어져야 한다.
수마트립탄
24명의 건강한 지원자(남성 14명, 여성 10명)에서 토피라메이트(12시간마다 100mg)의 반복 투여는 단회 투여의 수마트립탄의 경구(100mg) 또는 피하 투여(6mg)의 약물동력학에 영향을 미치지 않았다.
리스페리돈
토피라메이트를 100, 250, 400mg/day 로 용량을 증가하여 병용투여시 리스페리돈의 전신 노출이 감소되었다(250, 400mg/day에서 각각 정상상태 AUC의 16%, 33% 감소). 9‑히드록시히스페리돈 수치의 변화는 관찰되지 않았다. 리스페리돈과 토피라메이트 400 mg/day 병용투여 시 토피라메이트의 Cmax가 14% 증가하였고, AUC12가 12% 증가했다. 리스페리돈과 9‑히드록시히스페리돈 또는 토피라메이트의 전신 노출에 대해 임상적으로 유의한 변화는 없었으므로, 이 상호작용은 임상적으로 유의하지 않을 것으로 예상된다.
프로프라놀롤
34명의 건강한 지원자(남성 17명, 여성 17명)에 대한 토피라메이트(200mg/day) 반복 투여는 일일 160mg 용량의 프로프라놀롤 약물동력학에 영향을 미치지 않았다. 39명의 지원자(남성 27명, 여성 12명)에서 160mg/day의 프로프라놀롤 복용량은 200mg/day 용량의 토피라메이트 노출에 아무런 영향을 미치지 않았다.
디히드로에르고타민
24명의 건강한 지원자(남성 12명, 여성 12명)에서 토피라메이트(200mg/day)의 반복 투여는 1mg 피하 투여량의 디히드로에르고타민 약물동력학에 영향을 미치지 않았다. 유사하게, 디히드로에르고타민의 1 mg 피하 투여량은 동일한 연구에서 토피라메이트의 200 mg/day를 투여했을 때의 약물동력학에 영향을 미치지 않았다.
딜티아젬
토피라메이트(150mg/day)와 딜티아젬(240mg)을 병용투여 시, 딜티아젬 Cmax가 10% 감소하고 AUC가 25% 감소했으며, des‑acetyl diltiazem의 AUC는 18%, Cmax가 27% 감소하고, N‑desmethyl diltiazem에는 영향을 미치지 않았다. 토피라메이트와 딜티아젬의 병용투여는 결과적으로 토피라메이트의 Cmax를 16% 증가시키고, AUC12를 19% 증가시킨다.
벤라팍신
건강한 지원자에서 토피라메이트(150mg/day)를 반복투여 시 벤라팍신 또는 O‑desmethyl 벤라팍신의 약물동력학에 영향을 미치지 않았다. 벤라팍신(150 mg 서방제제)의 반복 투여는 토피라메이트의 약물동력학에 영향을 미치지 않았다.
3) 독성시험 정보
⓵ 발암성, 변이원성, 수태능이상
펜터민/토피라메이트
펜터민/토피라메이트 복합제로 발암성, 변이원성 또는 수태능이상을 평가한 동물 실험은 없었다. 다음 데이터는 이 약의 두 가지 주성분인 펜터민 또는 토피라메이트로 개별적으로 수행한 연구 결과를 바탕으로 한다.
펜터민
펜터민은 Ames 박테리아 변이원성 시험, Chinese 햄스터 폐 세포로 시행한 염색체 이상 분석 또는 생체내 소핵 분석에서 대사 활성화 또는 대사 증진이 없는 변이원성 또는 염색체성 물질이 아니다.
랫드에게 3, 10, 30 mg/kg/day의 펜터민을 2년간 경구 투여하였다. AUC 노출에 근거하여, 이 약 15mg/92mg의 최대 권장 투여량의 약 11~15배인 최고 용량의 펜터민(30 mg/kg)에서 발암성에 대한 증거는 없었다.
수태능이상에 대한 잠재력을 결정하기 위해 펜터민으로 수행한 동물실험은 없었다.
토피라메이트
토피라메이트는 일련의 시험관내/생체내 분석 시험에서 유전 독성 잠재력을 보이지 않았다. 토피라메이트는 Ames 시험이나 시험관내 마우스 림프종 시험에서 돌연변이성을 갖지 않았다. 그것은 시험관내에서 랫드의 간세포에서 부정기 DNA 합성(unscheduled DNA synthesis)을 증가시키지 않았으며, 시험관 내 인간 림프구나 생체 내 랫드의 골수에서 염색체 이상을 증가시키지 않았다.
21개월 동안 식사를 통해 토피라메이트(20, 75 및 300 mg/kg)를 투여한 마우스에서 방광 종양의 증가가 관찰되었다. 300 mg/kg을 투여 받은 암컷과 수컷에서 통계적으로 유의한 방광 종양 발생률의 상승은 주로 마우스에 조직 형태학적으로 고유한 평활근 종양의 증가로 인한 것이다. 300 mg/kg을 투여 받은 마우스의 혈장 노출은 이 약 15 mg/92 mg의 인체최대권장용량인 토피라메이트 단일요법을 받은 환자에서 측정한 정상 상태 노출의 약 2 ~ 4배이었다. 인체 발암성에 대한 이러한 발견의 관련성은 아직은 불확실하다. 최대 120 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 약 4 ~ 10배)까지의 용량으로 2년간 토피라메이트를 경구 투여한 결과 랫드에서 발암성의 증거는 발견되지 않았다.
최대 100mg/kg 또는 AUC에 근거한 이 약의 남성 및 여성에 대한 인체최대권장용량의 약 4 ~ 8배 노출 시 랫드에서 암수컷의 수태능에 영향이 없었다.
⓶ 생식발생독성시험
토피라메이트
이 약 성분인 토피라메이트는 여러 동물 종에서의 임상적으로 적절한 용량에서 최기형성을 비롯한 발생 독성을 유발한다.
기관형성기에 임신한 마우스에게 20, 100, 500 mg/kg으로 경구 투여했을 때, 모든 투여량에서 태아 기형 초기 두개안면결손) 발생률이 증가했다. 이 연구에서 토피라메이트 저용량(20 mg/kg)은 이 약의 15 mg/92 mg에서 mg/m2기준으로 토피라메이트의 인체최대권장용량의 약 2배이다. 태아의 체중과 골격골형성은 모체 체중 증가의 감소와 함께 500 mg/kg으로 감소되었다.
랫드 연구 (경구 투여량 20, 100, 500 mg/kg 또는 0.2, 2.5, 30, 400 mg/kg)에서 사지기형 (손발가락결손증 (ectrodactyly), 사지단소증 (micromelia), 무지증 (amelia))의 빈도는 임신 중 기관형성기간 동안 400 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량 34배) 이상으로 치료한 암컷의 자손사이에서 증가했다. 배아독성(태아 체중 감소, 구조적 변화의 발생률 증가)은 20 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량 2배) 정도의 낮은 용량에서 관찰되었다. 모체 독성에 대한 임상적 징후는 400 mg/kg 이상에서 나타나고, 모체의 체중 증가는 100 mg/kg 혹은 그 용량 이상으로 치료하는 동안 감소했다.
토끼 연구(기관형성기간 동안 경구로 20, 60, 180 mg/kg 또는 10, 35, 120 mg/kg)에서는 배/태아 사망률이 35 mg/kg(AUC 추정치에 근거한 인체최대권장용량의 2배)이상에서 증가였고, 최기형성의 영향(주로 늑골 및 척추 기형)이 120 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 6배)에서 관찰되었다. 모체 독성(체중 증가의 감소, 임상적 증후 및/또는 사망률)의 증거가 35 mg/kg 이상에서 나타났다.
임신 후반기와 수유기(0.2, 4, 20, 100 mg/kg 또는 2, 20, 200 mg/kg)의 암컷 랫드에 투여했을 때, 자손은 200 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 16배)에서 생존력이 감소하고 신체 발달이 지연되었으며, 2 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 2배) 이상에서 이유기 전 및/또는 후에 체중 증가가 감소하는 것으로 나타났다. 모체 독성(체중 증가의 감소, 임상 징후)은 100 mg/kg 혹은 그 이상에서 나타났다.
산후 성분(기관형성 중 0.2, 2.5, 30, 400 mg/kg)을 갖은 랫드의 배아/태아 발생 연구에서, 자손은 400 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 34배)에서 신체 발달이 지연되었고, 30 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 2배)이상에서 체중 증가의 지속적인 감소가 나타났다.
⓷ 임상시험 정보
감소된 칼로리 섭취 및 늘어난 신체 활동과 관련하여 체중 감량에 대한 이 약의 효과는 2개의 무작위배정, 이중맹검, 위약 대조 임상시험에서 비만 환자군(시험 1)와 비만 혹은 2가지 이상의 유의한 동반질환을 갖는 과체중 환자군(시험 2)에서 연구되었다. 두 임상시험 모두 4주간의 적정 기간이후 52주간의 치료 기간이 있었다. 치료 1년 후(56 주)에 측정된 2가지의 공동 1차 유효성 결과지표는 1) 기저치로부터의 체중 감소 백분율과 2) 기저치로부터 최소한 5%의 체중 감소 달성으로 정의되는 치료에의 반응이었다.
시험 1에서 비만 환자군(BMI 35kg/m2이상)는 1년 동안 2:1:2의 비율로 514명은 위약을, 241명은 이 약 3.75mg/23mg을, 512명은 이 약 15mg/92mg을 무작위로 처방 받았다. 환자는 18 ~ 71세(평균 연령 43세)로 구성되었으며, 83%가 여성이었다. 약 80%는 백인, 18%는 아프리카 계 미국인, 15%는 히스패닉/라틴계였다. 임상시험 시작 당시 환자의 평균 체중과 BMI는 각각 116kg과 42kg/m2였다. 제2형 당뇨 환자는 시험 1 참여에서 제외되었다. 이 임상시험 동안에 모든 환자에게 칼로리 섭취량이 약 500 kcal/day 감소하도록 균형 잡힌 저칼로리 식이 요법을 권장했으며, 영양 및 생활습관의 변화에 관한 상담을 제공했다.
시험 2에서 과체중 및 비만 환자군은 1년 동안 2:1:2의 비율로 994명은 위약을, 498명은 이 약 7.5mg/46mg을, 995명은 이 약 15mg/92mg을 무작위로 처방받았다. 임상시험에 적합한 환자는 27kg/m2 이상 45kg/m2 이하의 BMI(제2형 당뇨 환자의 경우 BMI에 대한 하한 값은 없음)와 다음의 비만관련 합병증에서 2가지 이상의 조건을 충족해야 했다.
• 고혈압(140/90 mmHg 이상 또는 당뇨인 경우 130/85 mmHg 이상) 또는 2가지 이상의 혈압강하제가 필요한 환자
• 200‑400 mg/dL 이상의 트리글리세리드 또는 2개 이상의 지질 저하 약물로 치료를 받고 있는 환자
• 공복 혈당 상승(100 mg/dL 이상) 또는 당뇨병;
• 허리 둘레: 남성 102cm 이상, 여성 88cm 이상
환자는 19 ~ 71세(평균 연령 51세)로 구성되었으며, 70%가 여성이었다. 약 86%는 백인, 12%는 아프리카 계 미국인, 13%는 히스패닉/라틴계였다. 임상시험 시작 당시 환자의 평균 체중과 BMI는 각각 103kg과 36.6kg/m2였다. 임상시험 시작 시 환자의 약 절반(53%)이 고혈압을 앓았다. 연구 시작 당시 제2형 당뇨 환자는 388명 (16%)이었다. 이 연구 동안에 모든 환자에게 칼로리 섭취량이 약 500 kcal/day 감소하도록 균형 잡힌 저칼로리 식이 요법을 권장했으며, 환자에게 영양 및 생활습관의 변화에 관한 상담을 제공했다.
무작위 배정된 환자 중 시험 1에서 40%, 시험 2에서 31%에 해당하는 상당수의 피험자가 56주 전에 연구를 중단하였다.
표 9은 시험 1과 시험 2에서 1년간의 체중 감량 결과를 나타낸다. 이 약 치료 1년 후, 모든 용량에서 위약에 비해 통계적으로 유의한 체중 감량을 초래했다 (표 9, 그림 1 및 2). 위약군 대비 이 약에 무작위 배정된 환자들의 통계적으로 상당히 유의한 비율에서 5% 및 10%의 체중감량을 달성하였다.
표 9. 시험 1과 시험 2에서 1년간의 체중 감량
그림 1. 시험 1 체중 변화 비율 그림 2. 시험 2 체중 변화 비율
시험 1과 시험 2의 비만과 관련된 심혈관, 대사 및 인체 계측 위험 요소의 변화는 표 10와 표 11에 나타내었다.
이 약을 사용한 1년간의 치료로 심박수를 제외하고 비만과 관련된 여러 위험 요소에서 위약보다 상대적으로 호전이 있었다.
표 10. 시험 1(비만)에서 1년간의 치료 후 위험 인자에 대한 기저치로부터의 최소 제곱근 평균† 변화(Least‑Squares (LS) Mean Change) 및 위약군과의 치료 결과 차이
시험 2에서 치료한 제2형 당뇨가 있는 388명의 대상자 중 기저치 대비 HbA1c 감소(6.8%)는 위약군에서는 0.1% 감소한 반면, 이 약 7.5 mg/46 mg 군 및 15 mg/92 mg군에서는 각각 0.4%씩 감소했다.
1) 이 약은 전문의약품, 일반의약품, 생약제제를 포함하여 다른 식욕억제제와 병용하여 사용하여서는 안된다. 이 약은 체중감량을 목적으로 투여하는 다른 의약품과의 병용투여에 대한 안전성▪유효성은 확립되지 않았다. 따라서, 이 약과 체중조절 목적으로 하는 다른 약물과의 병용은 권장되지 않는다.
2) 태아 독성 : 이 약은 태아에 유해한 영향을 줄 수 있다. 임신 및 역학 관련 연구 자료에 따르면 임신 초기에 이 약의 성분인 토피라메이트에 노출된 태아는 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다.
환자가 임신 중에 이 약을 복용하거나 이 약 복용 중에 임신한 경우, 치료를 즉시 중단하고 태아에게 미칠 잠재적인 위험이 있음을 환자에게 알려야 한다.
가임기 여성은 이 약으로 치료를 시작하기 전에 임신 테스트의 결과가 음성임을 확인해야 하고, 이 약으로 치료하는 동안에는 매달 한 번씩 임신 테스트를 해야 한다. 이를 위해 1회 1개월분 이상의 약을 처방하지 않아야 한다. 가임기 여성은 이 약으로 치료 중에는 효과적인 방법으로 피임을 해야 한다. [임부 및 수유부에 대한 투여 (7.1) 및 (7.4) 참조]
3) 이 약의 성분인 토피라메이트를 투여 받은 환자에서 중대한 피부반응(스티븐스존슨 증후군(SJS) 및 독성표피괴사용해(TEN))이 보고되었다. 대부분의 경우 스티븐슨존슨 증후군(SJS) 및 독성표피괴사용해(TEN)와 연관이 있다고 알려져 있는 다른 약물들을 함께 복용한 환자에서 발생하였으나, 몇몇 사례는 토피라메이트를 단독으로 복용하는 환자에서도 보고되었다. 따라서 중대한 피부반응의 징후를 환자에게 알리는 것이 권고된다. 스티븐슨존슨 증후군(SJS) 또는 독성표피괴사용해(TEN)가 의심될 경우, 이 약의 사용을 중단해야 한다.
2. 다음 환자에는 투여하지 말 것
1) 임신부
2) 녹내장 환자
3) 갑상선 기능 항진증 환자
4) MAO 억제제를 복용중이거나 또는 복용 후 14일이 경과하지 않은 환자 (혈압 상승 위험 유발)
5) 교감신경흥분성 아민에 대한 과민 반응 또는 특이체질 환자
6) 이 약의 성분에 과민증이 있는 환자
7) 18세 미만의 소아
8) 진전된 동맥경화증 환자
9) 심혈관계 질환 환자
10) 중등도 ~ 중증의 고혈압 환자
11) 폐동맥 고혈압 환자
12) 정신적으로 매우 불안하거나 흥분상태에 있는 환자
13) 약물남용의 병력이 있는 환자
3. 다음 환자에는 신중히 투여할 것
1) 신장애 환자
2) 간장애 환자
3) 이 약은 황색4호(타르트라진)를 함유하고 있으므로 이 성분에 과민하거나 알레르기 병력이 있는 환자에는 신중히 투여한다.(큐시미아캡슐7.5mg/46mg, 큐시미아캡슐11.25mg/69mg, 큐시미아캡슐15mg/92mg에 한함)
4) 이 약은 황색5호(선셋옐로우 FCF, Sunset Yellow FCF)를 함유하고 있으므로 이 성분에 과민하거나 알레르기 병력이 있는 환자에는 신중히 투여한다.(큐시미아캡슐7.5mg/46mg, 큐시미아캡슐11.25mg/69mg, 큐시미아캡슐15mg/92mg에 한함)
4. 이상반응
1) 임상시험
임상시험은 광범위하고 다양한 조건에서 시행되므로, 약물의 임상시험에서 관찰되는 이상반응 비율을 다른 약물의 임상시험에서 관찰되는 이상반응 비율과 직접적으로 비교할 수 없으며, 실제로 관찰되는 비율에 반영되지 않을 수도 있다.
이 자료는 평균 298일 동안 노출된 2,318명의 성인 환자(고혈압 환자 936명[40.4%], 제2형 당뇨 환자 309명[13.3%], BMI가 40 kg/m2 초과인 환자 808명[34.9%])를 대상으로 한 2개의 1년, 무작위, 이중 맹검, 위약 대조, 다기관 임상시험과 2개의 2상 임상 보조 연구를 통해 이 약에 대한 노출을 반영하여 기술하였다.
흔한 이상반응 : 5% 이상, 위약에 비해 최소 1.5배 이상으로 나타나는 이상반응으로는 지각 이상, 어지러움, 미각이상, 불면증, 변비, 구강 건조 등이 있다.
이 약의 치료 환자군의 2% 이상 및 위약군에서 보다 빈번하게 보고된 이상 반응은 표 1에 나와 있다.
표 1. 1년의 치료기간 동안 환자의 2% 이상 및 위약군에서 자주 보고된 이상 반응 ‑ 전반적인 연구 개체군 (Overall Study Population)
|
기관계 대분류(System Organ Class, SOC) 대표 용어(Preferred Term, PT) |
위약 (N=1561) % |
이 약 3.75mg/23mg (N=240) % |
이 약 7.5mg/46mg (N=498) % |
이 약 15mg/92mg (N=1580) % |
|
신경계 장애 |
|
|
|
|
|
지각 이상 |
1.9 |
4.2 |
13.7 |
19.9 |
|
두통 |
9.3 |
10.4 |
7.0 |
10.6 |
|
어지러움 |
3.4 |
2.9 |
7.2 |
8.6 |
|
미각이상 |
1.1 |
1.3 |
7.4 |
9.4 |
|
감각저하 |
1.2 |
0.8 |
3.6 |
3.7 |
|
주의력 장애 |
0.6 |
0.4 |
2.0 |
3.5 |
|
정신 장애 |
|
|
|
|
|
불면증 |
4.7 |
5.0 |
5.8 |
9.4 |
|
우울증 |
2.2 |
3.3 |
2.8 |
4.3 |
|
불안 |
1.9 |
2.9 |
1.8 |
4.1 |
|
위장관계 장애 |
|
|
|
|
|
변비 |
6.1 |
7.9 |
15.1 |
16.1 |
|
구강건조 |
2.8 |
6.7 |
13.5 |
19.1 |
|
오심 |
4.4 |
5.8 |
3.6 |
7.2 |
|
설사 |
4.9 |
5.0 |
6.4 |
5.6 |
|
소화불량 |
1.7 |
2.1 |
2.2 |
2.8 |
|
위-식도 역류 질환 |
1.3 |
0.8 |
3.2 |
2.6 |
|
구강 지각 이상 |
0.3 |
0.4 |
0.6 |
2.2 |
|
전신질환 및 투여 부위 상태 |
|
|
|
|
|
피로 |
4.3 |
5.0 |
4.4 |
5.9 |
|
과민성 |
0.7 |
1.7 |
2.6 |
3.7 |
|
갈증 |
0.7 |
2.1 |
1.8 |
2.0 |
|
가슴 불편함 |
0.4 |
2.1 |
0.2 |
0.9 |
|
시각 장애 |
|
|
|
|
|
시야흐림 |
3.5 |
6.3 |
4.0 |
5.4 |
|
안통 |
1.4 |
2.1 |
2.2 |
2.2 |
|
눈 건조 |
0.8 |
0.8 |
1.4 |
2.5 |
|
심장 장애 |
|
|
|
|
|
두근거림 |
0.8 |
0.8 |
2.4 |
1.7 |
|
피부 및 피하조직 장애 |
|
|
|
|
|
발진 |
2.2 |
1.7 |
2.0 |
2.6 |
|
탈모 |
0.7 |
2.1 |
2.6 |
3.7 |
|
신진 대사와 영양 장애 |
|
|
|
|
|
저칼륨혈증 |
0.4 |
0.4 |
1.4 |
2.5 |
|
식욕 감소 |
0.6 |
2.1 |
1.8 |
1.5 |
|
생식 기관 및 유방 질환 |
|
|
|
|
|
월경통 |
0.2 |
2.1 |
0.4 |
0.8 |
|
감염 |
|
|
|
|
|
상기도감염 |
12.8 |
15.8 |
12.2 |
13.5 |
|
비인두염 |
8.0 |
12.5 |
10.6 |
9.4 |
|
부비동염 |
6.3 |
7.5 |
6.8 |
7.8 |
|
기관지염 |
4.2 |
6.7 |
4.4 |
5.4 |
|
인플루엔자 |
4.4 |
7.5 |
4.6 |
4.4 |
|
요로감염 |
3.6 |
3.3 |
5.2 |
5.2 |
|
위장염 |
2.2 |
0.8 |
2.2 |
2.5 |
|
근골격계 및 결합 조직 장애 |
|
|
|
|
|
등통증 |
5.1 |
5.4 |
5.6 |
6.6 |
|
사지 통증 |
2.8 |
2.1 |
3.0 |
3.0 |
|
근육연축 |
2.2 |
2.9 |
2.8 |
2.9 |
|
근육골격통증 |
1.2 |
0.8 |
3.0 |
1.6 |
|
경부통증 |
1.3 |
1.3 |
2.2 |
1.2 |
|
호흡기, 흉부 및 종격동 장애 |
|
|
|
|
|
기침 |
3.5 |
3.3 |
3.8 |
4.8 |
|
굴울혈 |
2.0 |
2.5 |
2.6 |
2.0 |
|
인후두 통증 |
2.0 |
2.5 |
1.2 |
2.3 |
|
비충혈 |
1.4 |
1.7 |
1.2 |
2.0 |
|
상해, 중독 및 시술 후 합병증 |
|
|
|
|
|
시술 후 통증 |
1.7 |
2.1 |
2.4 |
1.9 |
이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 및 위약을 복용한 환자에서 손, 발, 얼굴 따끔거림 등의 지각 이상이 각각 4.2%, 13.7%, 19.9%, 1.9% 발생했다.
미각이상은 금속성 맛(metallic taste)으로 특징되고, 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 투여군에서 각각 1.3%, 7.4%, 9.4% 발생했으며, 이는 위약 투여군에서 1.1% 발생했다. 이러한 증상 대부분은 약물 치료 후 첫 12주 이내에 발생했다. 그러나 일부 환자군에서는 치료하는 동안 나중에 발생하는 것으로 보고되었다. 이 약 치료군에 해당하는 환자의 일부가 이러한 증상으로 인해 치료를 중단했다. (지각 이상의 경우 1%, 미각이상의 경우 0.6%).
기분 장애 및 수면 장애
이 약의 1년간 위약‑대조임상 시험에서 기분 장애 및 수면 장애와 관련된 하나 이상의 이상반응을 보고한 환자의 비율은 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 용량에서 각각 15.8%, 14.5%, 20.6%이었으며, 위약의 경우 10.3%였다. 이 증상들은 수면 장애, 불안 및 우울증으로 분류되었다. 수면 장애에 대한 보고는 일반적으로 불면증의 특징을 보였고, 이 약 3.75mg/23mg, 7.5mg/46mg 및 15mg/92mg 용량으로 치료받은 환자군에서 각각 6.7%, 8.1%, 11.1% 발생했으며, 위약으로 치료받은 환자군의 5.8%에서 발생했다. 불안은 이 약 3.75mg/23mg, 7.5mg/46mg, 15mg/92mg 용량으로 치료받은 환자군에서 각각 4.6%, 4.8%, 7.9% 발생했으며, 위약으로 치료받은 환자군의 2.6%에서 발생했다. 우울증/기분 장애에 대한 보고는 이 약 3.75mg/23mg, 7.5mg/46mg 및 15mg/92mg 용량으로 치료받은 환자군에서 각각 5.0%, 3.8% 및 7.6% 발생했으며, 위약으로 치료받은 환자군에서 3.4% 발생했다. 이러한 증상 대부분은 약물 치료 후 첫 12주 이내에 발생했다. 그러나 일부 환자군에서는 치료 과정에서 나중에 발생하는 것으로 보고되었다. 이 약의 임상 시험에서 기분과 수면 이상반응의 전반적인 유병률은 우울증 병력이 없는 환자에 비해 우울증의 병력이 있는 환자군에서 약 2배나 높았다. 그러나 기분 및 수면 이상반응이 나타난 환자들의 비율은 이 약으로 치료를 받는 환자군과 위약으로 치료를 받은 환자군에서 비슷하게 나타났다. 우울증 관련 증상은 모든 치료군에서 우울증 과거력이 있는 환자에서 더 빈번하게 발생했다. 그러나 이러한 증상의 발생률에 대한 위약‑보정된 차이(placebo‑adjusted difference)는 과거 우울증 병력에 관계없이 집단 간에 일정하게 유지되었다.
인지 장애
1년간의 위약‑대조임상 시험에서 1개 이상의 인지 관련 이상반응을 겪은 환자의 비율은 3.75mg/23mg 용량에서 2.1%, 7.5mg/46mg 용량에서 5.0%, 15mg/92mg 용량에서 7.6%, 위약에서 1.5% 였다. 이러한 이상반응은 주로 주의/집중력, 기억력, 언어(단어 선택) 문제였다. 이 증상들은 일반적으로 치료 첫 4주 이내에 시작되었고, 평균적으로 약 28일 이하의 기간 동안 지속되었으며, 치료를 중단하면 원상태로 회복되었다. 그러나 특정 환자의 경우 치료 후반 및 장기간동안 이 증상들을 경험했다.
실험실검사수치 이상
⓵ 혈청 중탄산염
1년간의 위약‑대조임상 시험에서 정상 범위 이하(2회 연속 방문 시 또는 최종 방문 시 21 mEq/L 미만의 수준)의 혈청 중탄산염의 지속적인 treatment‑emergent(TE) 감소 발생률은 3.75mg/23mg 투여군에서 8.8%, 7.5mg/46mg 투여군에서 6.4%, 15mg/92mg 투여군에서 12.8%, 위약 투여군에서 2.1%였다. 지속적이고 현저하게 낮은 혈청 중탄산염 수치(2회 연속 방문 시 또는 최종 방문 시 17 mEq/L 미만의 수준) 발생률은 3.75mg/23mg 투여군에서 1.3%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.7%, 위약 투여군에서 0.1% 였다. 일반적으로 혈청 중탄산염 농도의 감소는 경도 수준(평균 1‑3 mEq/L)이었고 치료 초기(4주 방문)에 발생했지만, 치료 후기에 중증 감소(severe decreases) 및 감소(decreases)가 발생하기도 했다.
⓶ 혈청 칼륨
1년간의 위약‑대조임상시험 중 지속적으로 낮은 혈청칼륨 수치(2회 연속 방문 시 또는 최종 방문 시 3.5 mEq/L 미만)의 발생률은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 3.6%, 15mg/92mg 투여군에서 4.9%, 위약 투여군에서 1.1%였다. 지속적으로 낮은 혈청칼륨(low serum potassium)을 경험한 피험자들 중 88%는 비‑칼륨보존이뇨제 (non‑potassium sparing diuretic)로 치료를 받았다.
임상시험기간 동안 현저히 낮은 혈청칼륨(3 mEq/L 미만, 치료 전보다 0.5 mEq/L 이상 감소)의 발생률은 3.75mg/23mg 투여군에서 0.0%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.7%, 위약 투여군에서 0.0%였다. 지속적으로 현저히 낮은 혈청칼륨(3 mEq/L 미만 및 2회 연속 방문시 또는 최종 방문시 치료 전보다 0.5 mEq/L 초과 감소)의 발생률은 3.75mg/23mg 투여군에서 0.0%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 0.1%, 위약 투여군에서 0.0%였다.
저칼륨혈증은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 1.4%, 15mg/92mg 투여군에서 2.5%, 위약 투여군에서 0.4%였다. 혈중 칼륨 감소는 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 0.4%, 15mg/92mg 투여군에서 1.0%, 위약 투여군에서 0.0%였다.
⓷ 혈청 크레아티닌
1년간의 위약‑대조임상시험에서 기저치 대비 복용량과 관련된 증가가 나타났으며, 4주에서 8주 사이에 정점에 도달했다가 감소하였으나 1년간의 치료 기간 동안 기저치 이상으로 상승 유지되었다. 치료기간 동안 0.3 mg/dL 이상의 혈청 크레아티닌 증가가 발생할 확률은 3.75mg/23mg 투여군에서 2.1%, 7.5mg/46mg 투여군에서 7.2%, 15mg/92mg 투여군에서 8.4%, 위약 투여군에서 2.0%였다. 기저치 대비 50% 이상의 혈청 크레아티닌이 증가한 비율은 3.75mg/23mg 투여군에서 0.8%, 7.5mg/46mg 투여군에서 2.0%, 15mg/92mg 투여군에서 2.8%, 위약 투여군에서 0.6% 였다.
신석증
1년간의 위약‑대조임상시험에서 신장결석증 발생률은 3.75mg/23mg 투여군에서 0.4%, 7.5mg/46mg 투여군에서 0.2%, 15mg/92mg 투여군에서 1.2%, 위약 투여군에서 0.3% 였다.
이상반응에 따른 약물 중단
1년간의 위약‑대조 임상시험에서 3.75mg/23mg 투여군에서 11.6%, 7.5mg/46mg 투여군에서 11.6%, 15mg/92mg 투여군에서 17.4%, 위약 투여군에서 8.4%가 보고된 이상반응으로 인해 약물 치료를 중단했다. 치료중단을 야기한 가장 흔한 이상반응은 표 2에 나타난다.
표 2. 1% 이상에서 약물 치료 중단을 야기한 이상반응(1 year clinical trials)
|
약물치료 중단을 야기한 이상반응a |
위약 (N=1561) % |
이 약 3.75mg/23mg (N=240) % |
이 약 7.5mg/46mg (N=498) % |
이 약 15mg/92mg (N=1580) % |
|
시야흐림 |
0.5 |
2.1 |
0.8 |
0.7 |
|
두통 |
0.6 |
1.7 |
0.2 |
0.8 |
|
과민성 |
0.1 |
0.8 |
0.8 |
1.1 |
|
어지러움 |
0.2 |
0.4 |
1.2 |
0.8 |
|
지각 이상 |
0.0 |
0.4 |
1.0 |
1.1 |
|
불면 |
0.4 |
0.0 |
0.4 |
1.6 |
|
우울증 |
0.2 |
0.0 |
0.8 |
1.3 |
|
불안 |
0.3 |
0.0 |
0.2 |
1.1 |
|
a 치료군 중 어느 하나에서라도1% 이상의 환자가 치료를 중단한 경우 | ||||
미국에서 이 약의 성분인 펜터민과 토피라메이트의 시판후 사용에 대하여 다음과 같은 이상반응이 보고되었다. 이러한 이상반응은 인구수를 특정하기 어려운 모집단으로부터 자발적으로 보고되기 때문에 발생 빈도를 정확하게 예측하거나 약물 노출과의 인과 관계를 확립하는 것이 항상 가능하지는 않다.
큐시미아
정신 장애 : 자살 생각, 자살 행동
안과 질환 : 급성폐쇄각녹내장, 안압 상승
펜터민
알레르기 이상반응 : 두드러기
심혈관 이상반응 : 혈압 상승, 허혈성 사건
중추 신경계 이상반응 : 다행감, 정신증, 진전
생식 이상반응 : 성욕의 변화, 발기부전
토피라메이트
피부질환 : 수포 피부 반응(다형 홍반, 스티븐존슨 증후군, 독성표피괴사용해 포함), 천포창
위장 장애 : 췌장염
간 질환 : 간 실조(사망자 포함), 간염
대사 장애 : 고암모니아혈증, 저체온증
안과 질환 : 황반증
5. 일반적 주의
1) 심박수 증가
이 약은 안정시 심박수를 증가시킬 수 있다.
이 약으로 치료한 과체중 및 비만 성인과 위약으로 치료한 과체중 및 비만 성인을 비교하였을 때, 이 약 치료군에서 기저치에 비해 분당 심박수가 5, 10, 15 및 20회 (bpm) 이상 증가한 환자의 비율이 높았다.
표 3은 최대 1년간의 임상연구에서 심박수의 상승을 보이는 환자의 수와 백분율을 나타낸다.
표 3. 단일 시점을 기준으로 기저치 심박수에서 심박수가 증가한 환자의 수와 백분율
지난 6개월 동안 심근경색이나 뇌졸중 병력이 있는 환자, 생명을 위협하는 부정맥 또는 울혈성심부전증이 있는 환자와 같은 심장 및 뇌혈관 질환 환자에 대해 특히 이 약의 치료로 인한 심박수 상승의 임상적 의미는 분명하지 않다.
이 약을 복용하는 모든 환자, 특히 심장 질환이나 뇌혈관 질환이 있는 환자 또는 이 약의 복용을 시작하거나 늘릴 때 안정시 심박수를 정기적으로 측정할 것을 권장한다. 이 약은 불안정한 심장 질환이나 뇌혈관 질환이 있는 환자를 대상으로 연구를 진행하지 않았으므로 이 환자들에 대한 사용은 권장하지 않는다.
환자는 이 약의 치료기간 동안 휴식을 취할 때 두근거림이나 고동치는 느낌을 경험하는 경우, 이 사실을 의료진에게 알려야 한다. 이 약을 복용하는 동안 안정시 심박수가 지속적으로 증가하는 환자의 경우, 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
2) 자살 행동과 자살 생각
이 약의 성분인 토피라메이트를 포함한 항경련제(AED)는 이러한 약물을 복용하는 목적과 상관없이 약물을 복용하는 환자에게 자살 충동이나 자살 행동 위험을 증가시킨다. 이 약으로 치료받는 환자는 우울증의 발생이나 증상의 악화, 자살 충동이나 자살 행동 및/또는 환자의 기분이나 행동의 비정상적인 변화에 대해 모니터링되어야 하며, 이러한 증상 또는 행동이 발현될 경우 즉시 의료전문가에게 보고될 수 있도록 한다. 자살 충동이나 자살 행동을 경험한 환자는 이 약의 복용을 중단한다.
자살 시도나 적극적인 자살 생각의 병력이 있는 환자는 이 약의 복용을 피해야 한다.
여러 적응증이 있는 11가지 항경련제에 대한 199개의 위약 대조 임상연구(단일요법 및 보조요법, 중간 치료 기간 12주)를 통합 분석한 결과, 항경련제 중 하나에 무작위로 배정된 환자는 자살 충동이나 행동이 위약에 무작위로 배정된 환자에 비해 약 2배의 위험도(보정 상대 위험도(adjusted relative risk) 1.8, 95% 신뢰 구간 [CI] 1.2, 2.7)를 나타냈다. 위약으로 치료한 환자 16,029명에서의 자살 행동이나 자살 생각의 예상 발생률은 0.24%인 것에 비해, 항경련제로 치료한 환자 27,863명에서 예상 발생률은 0.43%로, 치료받은 환자 530명당 약 1건의 자살 생각이나 행동의 증가가 나타났다. 임상시험에서 항경련제로 치료받은 환자에서는 4건의 자살이 있었고 위약으로 치료받은 환자에서는 한 건도 없었지만, 그 수가 너무 적어 자살과 관련된 항경련제 영향에 대해서는 결론을 내릴 수 없었다.
항경련제로 인한 자살 충동이나 행동 위험의 증가는 항경련제로 약물 치료를 시작한 후 빠르면 1주일 후에 관찰되었으며, 치료 기간 내내 지속되었다. 임상분석에 포함된 대부분의 임상시험들은 24주를 초과하지 않았으므로, 24주 이후의 자살 충동이나 행동의 위험을 평가할 수 없었다.
자살 충동이나 행동의 위험은 분석된 데이터에 사용된 약물에서 일반적으로 일치했다. 다양한 작용 기전과 다양한 범위의 적응증을 가진 항경련제에서 위험 증가가 확인되었다는 것은, 항경련제의 적응증과는 무관하게 모든 항경련제에 위험성이 있다는 것을 시사한다. 위험도는 분석된 임상시험에서 연령(5세에서 100세)에 따라 크게 변하지 않았다.
3) 급성 근시 및 2차성 폐쇄각녹내장
이 약의 성분인 토피라메이트로 치료받은 환자들에서 2차성 폐쇄각녹내장과 관련된 급성 근시 증후군이 보고되었으며, 그 증상에는 갑작스런 시력저하 및/또는 안통이 포함된다. 안과적인 검사시 근시, 좁아진 전방, 안 충혈 및 안압 상승이 관찰될 수 있다. 산동은 나타날 수도 있고 아닐 수도 있다. 이 증후군은 모양체상방 삼출 (supraciliary effusion)로 인한 수정체와 홍체의 전방 이동 및 2차성 폐쇄각녹내장과 관련이 있을 수 있다. 증상은 일반적으로 토피라메이트 치료 시작 후 1개월 이내에 발생하지만 치료 중 언제든지 발생할 수 있다. 증상에서 회복되기 위한 1차 치료는 이 약의 복용을 즉각 중단하는 것이다.
어떤 병에 의한 안압상승이라도 치료받지 않으면 영구 시력손실을 포함한 심각한 후유증을 가져올 수 있다.
이 약의 성분인 토피라메이트를 투여 받은 환자에게서 안압 상승과 독립적으로 시야결손이 보고되었다. 임상시험에서 이러한 반응의 대부분은 토피라메이트 중단 후 가역적이었다. 만일 이 약 치료 중 어느 때라도 시각 문제가 발생한다면 약물 중단을 고려해야 한다.
4) 기분 장애 및 수면 장애
이 약은 우울증과 불안, 불면증 등 기분 장애를 유발할 수 있다. 우울증 병력이 있는 환자는 이 약을 복용하는 동안 우울증 또는 기타 기분 장애의 재발 위험이 높아질 수 있다. 이 기분 장애와 수면 장애의 대부분은 자연적으로 치료되었거나 복용 중단 시 치료되었다. 임상적으로 유의미하거나 지속적인 증상이 나타나는 경우, 이 약의 복용량을 줄이거나 복용 중단을 고려해야 한다. 환자에게서 자살 생각이나 자살 행동 증상을 보이면 이 약의 복용을 중단해야 한다.
5) 인지 장애
이 약은 인지 기능 장애(예: 주의/집중력 장애, 기억곤란, 언어 장애, 특히 단어 선택의 어려움)을 유발할 수 있다.
이 약의 용량을 빠르게 적정하거나 초기에 고용량으로 복용하면 주의력, 기억력, 언어/단어 선택 어려움과 같은 인지기능 관련 이상반응 발생률을 높이는 경우가 있다.
이 약은 인지 기능을 손상시킬 가능성이 있기 때문에, 환자는 이 약의 치료가 악영향을 미치지 않는다고 확신할 수 있을 때까지 자동차를 포함한 위험한 기계를 조작할 때 주의하여야 한다. 인지 기능 장애가 지속되면 감량을 고려해야 하며, 중등도에서 중증까지의 증상, 불편함 또는 복용량 감소로 해결하지 못한 증상에 대해 이 약의 복용량 감소 또는 복용 중단을 고려해야 한다.
6) 대사성산증
이 약으로 치료한 환자에서 음이온 차의 변화 없는(non‑anion gap) 고염소혈증성 대사성 산증(만성 호흡성 알칼리혈증 없이 정상 범위 이하로 혈청 중탄산염 농도 저하)이 보고되었다. 신장질환, 심한 호흡기질환, 간질 중첩증, 설사, 수술, 케톤 식이 등 산증의 위험을 증가시킬 수 있는 상태 혹은 치료가 토피라메이트의 중탄산염 농도 저하 효과를 증가시킬 수 있다. 이 약과 탄산탈수효소억제제(예: 조니사미드, 아세타졸아미드 또는 디클로펜아미드)를 병용할 경우 대사성산증의 중증도를 증가시킬 수 있으며 또한 신장 결석 형성의 위험도도 높아질 수 있다. 따라서 대사성산증이 발생하기 쉬운 상태의 환자가 이 약을 다른 탄산탈수효소억제제와 병용하는 경우에는 대사성산증의 발생이나 악화 여부를 모니터링 하여야 한다.
급성 또는 만성 대사성산증의 일부 증상으로는 과다호흡, 피로 및 거식증과 같은 비특이적 증상, 또는 심장부정맥 또는 혼미를 포함한 보다 심각한 후유증이 있을 수 있다. 만성적이고 치료되지 않은 대사성산증은 신석증이나 신장석회증의 위험도를 높일 수 있으며, 골연화증(소아 환자에서 구루병이라고도 함) 및/또는 골절 위험이 높은 골다공증을 유발할 수 있다. 성장 및 뼈 관련 후유증에 대한 이 약의 효과는 장기적 위약 대조 임상 시험에서 체계적으로 조사되지 않았다.
이 약의 치료 전과 치료 중에 혈청 중탄산염을 포함한 전해질 측정이 권장된다. 이 약의 임상시험에서 혈청 중탄산염의 피크 감소는 4주째까지 발생했으며, 대부분 피험자에서는 시험약 변경 없이 56주째까지 중탄산염 교정이 있었다. 그러나 이 약의 복용 중 지속적인 대사산증이 발생하면 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
7) 크레아티닌 상승
이 약은 혈청 크레아티닌 증가를 유발할 수 있다. 4주에서 8주간의 치료 후에 혈청 크레아티닌의 피크 증가가 관찰되었다. 평균적으로 혈청 크레아티닌은 점차 감소했지만 기저치 크레아티닌 값보다 높게 유지되었다. 혈청 크레아티닌의 변화(측정된 GRF)는 치료를 중단하면 회복되지만, 만성적으로 치료시에 신장 기능에 미치는 영향에 대해서는 알려지지 않았다. 따라서 이 약 치료 시작 전과 치료 중에 혈청 크레아티닌 측정이 권장된다. 이 약의 복용 중에 크레아티닌의 지속적인 상승이 발생하면 이 약의 복용량을 줄이거나 복용을 중단해야 한다.
8) 당뇨병 치료를 하는 제2형 당뇨 환자에서 저혈당증의 잠재적 위험
체중 감량은 인슐린 및/또는 인슐린 분비 촉진제(예: 설폰요소제)로 치료되는 제2형 당뇨 환자의 저혈당증 위험을 높일 수 있다. 이 약은 인슐린과의 병용 치료에 관해서는 연구되지 않았다. 제2형 당뇨 환자에 대해서는 이 약의 치료 시작 전과 치료 중에 혈당 수치를 측정할 것을 권장한다. 저혈당의 위험을 감소시키기 위해서 비포도당 의존성(non‑glucose‑dependent) 항당뇨병제의 약물 복용량 감소를 고려해야 한다. 이 약의 복용을 시작한 후 환자가 저혈당증을 일으키면 당뇨병치료제 처방을 적절히 변경해야 한다. 또한, 이 약 투여 시 당뇨병 환자에서 인슐린 요구량이 변경될 수 있으므로 당뇨병 환자에게 식이요법과 병행하여 이 약을 투여할 경우 인슐린 투여량을 조절하여야 한다.
9) 혈압강하제로 치료받는 환자에게서 저혈압의 잠재적 위험
혈압강하제로 치료를 받는 고혈압 환자의 경우, 체중 감량은 저혈압의 위험을 높일 수 있으며 어지러움, 어질어질함, 실신 등을 유발할 수 있다. 고혈압 치료를 받는 환자는 이 약의 치료 시작 전과 치료 중 혈압 측정을 권장한다. 이 약의 복용을 시작한 후 환자에게서 저혈압과 관련된 증상을 보이면 항고혈압제 처방을 적절하게 변경해야 한다.
10) 알코올을 포함한 중추신경 억제제로 인한 중추신경기능저하
펜터민 또는 토피라메이트와 알코올 또는 중추신경 억제제(예: 바르비탈계, 벤조디아제핀 및 수면제)를 병용하면 어지러움, 인지기능 관련 이상반응, 나른함, 어질어질함, 조정력 저하 및 졸림과 같은 중추신경기능저하나 이러한 약제의 기타 중추 매개 효과를 증가시킬 수 있다. 따라서 이 약은 알코올을 함께 복용하면 안 된다.
11) 갑작스런 복용 중단으로 인한 잠재적 발작
이 약의 성분인 토피라메이트의 갑작스런 복용 중단으로 인한 금단 현상은 발작이나 간질 병력이 없는 개인에게도 발작 증상을 일으키는 경우가 있다. 이 약의 즉각적인 복용 중단이 의학적으로 요구되는 상황에서는 환자에 대한 적절한 모니터링이 권장된다. 이 약 15mg/92mg 용량의 복용을 중단하는 환자는 발작을 일으킬 가능성을 줄이기 위해 권장하는 대로 그 복용량을 점차적으로 줄여야 한다.
12) 신장애 환자
이 약의 성분인 펜터민과 토피라메이트는 신장 배설을 통해 제거된다. 따라서 중등증(크레아티닌 청소율(CrCl)이 30 mL/min 이상 50 mL/min 미만) 또는 중증(CrCl 30 mL/min 미만) 신장애 환자에서 펜터민과 토피라메이트에 대한 노출이 더 높다. 두 환자군에서 모두 이 약의 용량을 조절한다.
이 약은 신장 투석 중에 있는 말기 신장질환 환자에 대해서는 연구되지 않았다. 이 환자군에서는 이 약의 사용을 피해야 한다.
13) 간장애 환자
경증(Child‑Pugh score 5 ‑ 6) 또는 중등증(Child‑Pugh score 7 ‑ 9) 간장애 환자에서 펜터민에 대한 노출은 건강한 지원자에 비해 더 높았다. 중등증의 간장애 환자에서는 이 약의 용량을 조정한다.
이 약은 중증 간장애 환자(Child‑Pugh score 10 ‑ 15)에 대해서는 연구되지 않았다. 이 환자군에서는 이 약의 사용을 피해야 한다.
14) 신장 결석
이 약의 복용은 신장결석 형성과 관련이 있다. 이 약의 성분인 토피라메이트는 탄산탈수효소활성(carbonic anhydrase activity) 을 억제하고 뇨중 구연산의 배설을 감소시켜, 뇨의 pH를 높임으로써 신장 결석 형성을 촉진한다.
탄산탈수효소(예: 조니사미드, 아세타졸아미드, 메타졸아미드) 를 억제하는 다른 약물과 함께 이 약을 사용하지 않는다.
케톤식이요법(Ketogenic Diet)을 하는 환자의 토피라메이트 사용은 또한 신장 결석 형성의 가능성을 증가시키는 생리적 환경을 초래할 수 있다.
수분 섭취량을 늘려서 소변량을 늘리면 신장 결석 형성에 관련있는 물질의 농도를 낮출 수 있다. 운동이나 높은 기온에 노출되기 전 및 중에 적절한 수분 공급을 하는 것은 열과 관련된 이상반응의 위험을 줄일 수 있다
15) 땀분비 감소증 및 고체온증
이 약의 성분인 토피라메이트의 복용으로 입원이 필요할 수도 있는 땀분비 감소증이 보고되었다. 땀분비 감소와 정상보다 높은 체온 증가가 특징으로 나타난다. 일부 사례는 온도가 상승한 환경에 노출된 이후에 토피라메이트 복용과 함께 보고되었다.
이 약으로 치료받는 환자는 신체 활동 중, 특히 더운 날씨에 땀분비 감소와 체온 증가를 모니터링 해야 한다. 이 약을 열 관련 질환에 걸리기 쉬운 환자에게 다른 약제와 함께 처방할 때는 각별히 주의해야 한다. 이들 약제에는 다른 탄산탈수효소억제제 및 항콜린성 작용 약제가 포함되나 이에 한정되지는 않는다.
16) 저칼륨혈증
이 약은 탄산탈수효소활성의 억제를 통해 저칼륨혈증의 위험을 높일 수 있다. 또한 이 약을 푸로세미드(루프이뇨제) 또는 히드로클로로티아지드 (티아지드 유사 이뇨제)와 같은 비‑칼륨보존이뇨제(non‑potassium sparing diuretics)와 병용시 칼륨 손실을 더욱 증가시킬 수 있다. 이 약 처방 시 환자는 저칼륨혈증에 대해 모니터링되어야 한다.
17) 모니터링: 실험실적 검사
이 약은 무작위, 이중 맹검, 위약 대조 임상연구에서 여러 실험실적 분석 결과의 변화를 일으키는 경우가 있었다.
기저시점 및 치료 기간 중 주기적으로 중탄산염, 크레아티닌, 칼륨 및 혈당을 포함하는 혈액 화학 프로필을 확보해야 한다.
18) 남용
이 약의 주성분인 펜터민은 약물 남용의 가능성이 있다. 펜터민은 암페타민과 화학적 및 약리학적으로 관련이 있다. 암페타민과 기타 각성제는 광범위하게 남용되어 왔으며, 이 약을 체중 감량 프로그램의 일부로 포함시키는 것이 바람직하다고 평가할 때 펜터민의 남용 가능성을 염두에 두어야 한다. 암페타민 및 관련 약물(예: 펜터민)의 남용은 약물 사용 조절 장애와 심각한 사회적 기능 장애를 일으키는 경우가 있다. 이 약의 복용량을 권장 용량 보다 여러 번 늘린 환자에 대한 보고가 있다.
19) 의존성
이 약은 육체적 약물 의존 가능성에 대해 체계적으로 연구되지는 않았다. 육체적 약물 의존성은 반복되는 약물 복용에 대해서 생리적으로 적응하려는 상태이다. 육체적 약물 의존성은 약물 사용의 갑작스런 중단 또는 약물의 현저한 (복)용량 감소 이후에 약물 종류별 금단 현상으로 나타난다.
이 약의 각 주성분에 대한 육체적 약물 의존 가능성에 대한 정보는 한정되어 있다. 토피라메이트의 경우, 갑작스러운 약물 중단은 발작이나 간질 병력이 없는 환자에서도 발작이 일어나는 경우가 있었다. 펜터민의 경우, 장기적인 고용량 복용 후 갑작스런 중단은 극심한 피로와 정신적 우울증을 야기한다. 수면의 뇌파에도 변화가 나타난다. 따라서 이 약의 신속한 복용 중단이 필요한 상황에서는 적절한 의학적 모니터링이 권장된다.
20) 이 약은 최근 1년 이내에 다른 식욕억제제를 사용한 환자에게는 투여가 권장되지 않으므로 주의한다.
21) 토피라메이트를 투여 받은 환자에서 뇌병증을 동반 혹은 동반하지 않은 고암모니아혈증이 보고되었다. 고암모니아혈증은 토피라메이트와 발프로산을 병용 투여한 경우 더 빈번하게 보고되었다(상호작용 항 참조). 고암모니아혈증은 임상적으로 증상이 없을 수 있으나, 고암모니아혈증성 뇌병증은 기면 또는 구토를 동반한 의식수준 및/또는 인지기능에 있어서 급격한 변화를 자주 보인다. 이 약과 관련하여 설명할 수 없는 기면, 구토 혹은 정신상태의 변화가 발생한 환자의 경우, 고암모니아혈증성 뇌병증을 고려하여 혈중 암모니아 수치를 측정하는 것이 권장된다.
6. 상호작용
1) 모노아민산화효소억제제
고혈압 발생의 위험성 때문에 모노아민 산화효소 억제제(monoamine oxidase inhibitors) 투여기간 동안 또는 투여 후 14일 동안 펜터민 복용을 금한다.
2) 경구피임약
건강한 지원자와는 달리 비만환자에게서 에티닐에스트라디올(에스트로겐 성분) 35 ug과 노르에틴드론 (프로게스틴 성분) 1 mg을 함유한 경구 피임약의 단회 용량과 이 약 15 mg/92 mg의 1일 1회 병용 투여는 에티닐에스트라디올의 노출을 16% 감소시키고, 노르에틴드론의 노출을 22% 증가시켰다.
이 연구가 피임 효과에 대한 상호 작용의 영향을 구체적으로 다루지는 않았지만, 임신의 위험도가 증가할 것이라고 예상되지는 않는다. 피임 효과의 주된 결정 요인은 복합 경구 피임약의 프로게스틴 성분이므로 높아진 프로게스틴 노출이 유해할 것으로 예상되지는 않는다.
그러나 불규칙한 출혈(점상출혈)은 자궁 내막을 안정화시키는 프로게스틴에 대한 노출 증가와 에스트로겐 노출 감소로 인해 더 자주 발생할 수 있다. 환자는 출혈이 발견되면 복합 경구 피임약을 중단하지 말아야 한다는 사실을 통보 받아야 한다. 그러나 점상출혈이 불편할 경우 의사에게 알려야 한다.
3) 알코올을 포함한 중추신경억제제
이 약과 알코올 또는 기타 중추신경억제제과의 특정 약물상호작용에 관한 연구는 수행되지 않았다. 펜터민 또는 토피라메이트와 알코올 또는 중추신경억제제(예: 바르비탈류, 벤조디아제핀 및 수면제)의 병용 투여는 어지러움 또는 인지 이상반응 또는 이러한 약물의 중추 매개 효과와 같은 중추신경기능저하를 증가시킬 수 있다. 따라서 이 약을 알코올이나 다른 중추신경억제제와 함께 사용하는 경우 환자는 중추신경기능저하나 이상반응 위험이 증가할 가능성에 대해 상담받아야 한다.
4) 비‑칼륨보존이뇨제
비‑칼륨보존이뇨제와 함께 이 약을 병용투여하면 이러한 이뇨제의 칼륨 손실 작용이 증가할 수 있다. 토피라메이트 단일제와 히드로클로로티아지드 단일제를 병용하는 경우 토피라메이트의 Cmax 및 AUC가 각각 27% 및 29% 증가하는 것으로 나타났다. 비‑칼륨보존 의약품과 이 약을 병용 처방할 때, 환자의 저칼륨혈증에 대해 모니터링해야 한다.
5) 항경련제
간질 환자가 토피라메이트와 페니토인 또는 카르바마제핀을 병용하는 경우, 토피라메이트를 단독 복용하는 것에 비해 토피라메이트의 혈장 농도가 각각 48%와 40% 감소했다.
발프로산과 토피라메이트의 병용투여는 뇌병증과는 무관하게 고암모니아혈증을 일으키는 경우가 있다. 또한 발프로산과 토피라메이트를 병용한 환자에서 고암모니아혈증과 무관하게 저체온증이 나타나는 경우가 있다. 저체온증이나 뇌병증의 발병이 보고되는 경우, 환자의 혈중 암모니아 수치를 검사하는 것이 권장된다.
6) 탄산탈수효소억제제
이 약의 성분인 토피라메이트를 다른 탄산탈수효소억제제(예: 조니사미드, 아세타졸아미드,디클로펜아미드)와 함께 사용하면 대사성산증의 중증도가 증가할 수 있으며 신장 결석 형성의 위험도 증가할 수 있다. 이 약을 탄산탈수효소를 억제하는 다른 약물과 함께 사용해서는 안된다.
7) 피오글리타존
이 약의 성분인 토피라메이트와 피오글리타존의 병용투여 임상시험에서 피오글리타존과 활성대사산물 노출의 손실이 보고되었다. 이 결과의 임상적유의성은 알려지지 않았다. 그러나 피오글리타존 치료 중 이 약의 병용 또는 이 약 치료 중 피오글리타존 병용 시, 당뇨병 환자의 상태를 적절하게 통제하기 위해 환자에 대한 일상적인 모니터링 시 세심한 주의가 필요하다.
8) 아미트리프틸린
일부 환자는 이 약의 성분인 토피라메이트 복용 시 아미트리프틸린 농도가 크게 증가할 수 있다. 이 약과 병용투여 시 아미트리프틸린 용량 조절은 혈청 수치가 아닌 환자의 임상 반응에 따라 이루어져야 한다.
7. 임부 및 수유부에 대한 투여
1) 임부
이 약은 임부에게 사용해서는 안된다. 이 약은 태아에게 유해한 영향을 줄 수 있으며, 이 약 사용으로 인한 체중 감량은 임신한 여성에게 잠재적인 이득이 되지 않는다. 역학자료에서는 이 약의 성분인 토피라메이트에 임신 초기 3개월 동안 노출시 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다. 여러 종의 임신한 동물이 임상적으로 적절한 용량의 토피라메이트를 복용했을 때 두개 및 안면 결손을 포함한 구조적 기형 및 자궁 내 태아 체중 감소가 후손에서 발생했다.
환자가 임신 중에 이 약물을 복용하거나 이 약물 복용 중에 임신한 경우, 치료를 즉시 중단하고 태아에게 미칠 잠재적인 위험이 있음을 환자에게 알려야 한다.
임상적 고찰
구순구개 파열은 임신 5주부터 9주까지 발생한다. 입술은 임신 5주에서 7주 사이에 형성되며, 구개(입천장)는 임신 6주부터 9주 사이에 형성된다.
임신 중 모체 조직에서 발생하는 필수적인 체중 증가 때문에 이미 과체중이거나 비만인 임산부 뿐 아니라, 모든 임산부의 경우 최소한의 체중 증가 및 체중 감량이 되지 않는 것이 권장된다.
이 약은 대사성산증을 일으킬 수 있다. 토피라메이트 유발 대사성산증의 영향은 임신에 대해서는 연구되지 않았다. 그러나 다른 원인으로 인한 임신 중 대사성산증은 태아의 성장 감소, 태아에게 공급되는 산소 감소 및 태아 사망을 유발할 수 있으며, 분만을 견딜 수 있는 태아의 능력에 영향을 미칠 수 있다.
임상 데이터
임신 중에 이 약의 구성성분인 토피라메이트 노출로 인한 주요 선천성 기형 및 구순구개 파열 위험을 평가한 데이터는 North American Anti‑Epileptic Drug (NAAED) Pregnancy Registry 및 몇 가지 대규모 후향적 역학 연구에서 확인할 수 있다. NAAED Pregnancy Registry는 구순구개 파열 위험이 9.60(95% CI 3.60 ‑ 25.70)으로 증가할 것으로 예상했다. 대규모 후향적 역학 연구에서 임신 중 토피라메이트 단일요법 노출이 구순구개 파열 위험성의 약 2~5배 증가와 연관이 있었다. (표 4). FORTRESS 연구에서는 임신 초기에 토피라메이트에 노출된 1,000명의 유아 당 구순구개 파열 증상의 위험성이 1.5배 (95% CI = ‑1.1 ~ 4.1) 증가가 관찰되었다.
표 4. 자궁 노출 및 구순구개 파열 및 주요 선천성 기형에서의 토피라메이트 연관성 평가 연구 요약
동물 데이터
펜터민/토피라메이트
태아 발육 연구는 랫드와 토끼에서 펜터민과 토피라메이트 병용 요법으로 수행되었다. 기관이 형성되는 기간에 랫드에게 투여된 펜터민과 토피라메이트는 태아의 체중 감소를 가져 왔지만, 최고용량인 펜터민 3.75 mg/kg과 토피라메이트 25 mg/kg에서 태아 기형을 일으키지 않았다. (각 주성분에 대해 AUC 예상치에 근거한 인체최대권장용량의 약 2배). 토끼를 대상으로 한 유사한 연구에서는 AUC에 근거한 인체최대권장용량의 약 0.1배(펜터민) 및 1배(토피라메이트)의 임상 노출이 있었을 때, 태아 발달에 대한 어떠한 영향도 관찰되지 않았다. 랫드와 토끼에서 이 용량으로 유의미하게 낮은 모체의 체중 증가가 기록되었다.
산전/산후 발달 연구는 펜터민과 토피라메이트 치료의 병용 요법으로 랫드에서 시행되었다. 1.5 mg/kg/day의 펜터민과 10 mg/kg/day의 토피라메이트 (AUC 기준 인체최대권장용량에서 각각 약 2배와 3배의 임상 노출)로 기관형성에서 수유기까지 치료한 랫드의 모체 또는 자손에 대한 유해한 영향은 없었다. 고용량 11.25 mg/kg/day의 펜터민과 75 mg/kg/day의 토피라메이트(AUC 기준 최대 임상 용량의 약 5배와 6배)를 투여하면 모체 체중 증가의 감소와 자손 독성을 초래한다. 자손에 대한 영향으로는 출생 후 자손 생존률 감소, 사지 및 꼬리의 기형 증가, 자손 체중 감소 및 학습, 기억 또는 수정과 생식에는 영향을 미치지 않으며 성장, 발달 및 성적 성숙을 지연시켰다. 사지 및 꼬리 기형은 토피라메이트 단독으로 수행한 동물 연구의 결과와 일치했다.
펜터민
펜터민에 대한 동물 생식 연구는 수행되지 않았다. 펜터민/토피라메이트 병용 요법으로 실시한 랫드 대상 연구로 부터 제한된 데이터는 펜터민 단독으로는 최기형성을 보이지 않았지만 AUC 기준으로 이 약의 인체최대권장용량의 5배에서 저체중과 태아의 생존률 감소를 보였다.
토피라메이트
토피라메이트는 임상적으로 적절한 용량에서 최기형성을 포함한 발달 독성을 유발한다.
2) 분만
이 약이 인간의 분만에 미치는 영향은 알려지지 않았다. 산모 및/또는 태아에서 이 약으로 유도되는 대사성산증의 발달은 분만을 견딜 수 있는 태아의 능력에 영향을 미칠 수 있다.
3) 수유부
토피라메이트와 암페타민(펜터민은 암페타민과 유사한 약리학적 활성 및 화학적 구조를 가짐)이 모유에서 배출되기 때문에 이 약이 사람의 모유에 존재할 수 있다. 신생아의 모유 수유에 대한 심각한 이상반응의 잠재적 위험성때문에, 산모에 대한 약물 처방의 중요성을 고려하여 수유를 중단할 것인지 약물 투여를 중단할 것인지에 대한 여부를 결정해야 한다.
4) 가임여성
이 약은 태아에 유해할 수 있다. 임신 등록 및 역학 연구의 자료에 따르면 임신 초기에 이 약 성분인 토피라메이트에 노출된 태아는 구순구개 파열(구개파열을 동반 혹은 미동반한 구순파열)의 위험이 증가하는 것으로 나타난다. 이 약 치료 중 임신한 여성은 즉시 치료를 중단하고 의료진에게 알려야 한다.
임신 테스트
가임기 여성은 이 약의 치료를 시작하기 전에 임신 테스트의 결과가 음성임을 확인해야 하고, 이 약으로 치료하는 동안에는 매달 한 번씩 임신 테스트를 해야 한다.
피임
이 약 치료 중 가임 여성은 효과적인 피임법을 사용해야 한다.
8. 소아에 대한 투여
18세 미만 소아 환자를 대상으로 한 이 약의 안전성과 효과는 입증되지 않았으며, 이 약의 사용은 소아 환자에게 권장되지 않는다. 이 약의 성분인 토피라메이트를 복용한 소아 환자에서 나타나는 심각한 이상반응으로는 급성 녹내장, 땀분비 감소증 및 고체온증, 대사성산증, 인지 및 신경 정신적 반응, 고암모니아혈증 및 뇌병증, 신결석이 있다.
어린 동물 연구
이 약에 대한 어린 동물 연구는 수행되지 않았다. 어린 연령(출생 후 12 ~ 50일)의 랫드에게 토피라메이트(30, 90 또는 300 mg/kg/day)를 경구 투여했을 때, 가장 높은 용량에서 수컷 랫드의 뼈 성장판 두께가 감소했다.
9. 고령자에 대한 투여
이 약의 임상 시험에서 65세 이상 환자는 총 254명(7%)이었다. 고령 피험자와 젊은 연령의 피험자 사이에 안전성이나 효과에 전반적인 차이는 없었지만, 일부 고령자에게서 나타나는 더 큰 과민반응은 배제할 수 없었다.
이 약의 임상 연구에는 젊은 연령의 피험자와 다르게 반응하는지를 판단할 수 있을 정도의 충분한 65세 이상 피험자 수가 포함되지 않았다. 일반적으로 고령 환자에 대한 투여량 선택은 신중해야 하며, 투여량 선택은 보통 투여 범위의 최하위부터 시작하며, 간, 신장 또는 심장 기능 저하와 동반 질환 또는 기타 약물 치료의 빈도가 높아지는 것을 반영해야 한다.
10. 신장애 환자
건강한 지원자와 비교할 때 C‑G 공식(Cockcroft‑Gault equation)에 의해 평가된 중등증 및 중증 신장애 환자에서 펜터민과 토피라메이트에 대해 더 높은 노출을 보였다.
경증 신장애가 있는 환자에게는 용량 조절이 필요하지 않다. 중등증(30 이상 50 mL/min 미만의 크레아티닌 청소율[CrCl]) 및 중증 (30 mL/min 미만의 CrCl) 신장애 환자에게 있어 복용량은 1일 1회 7.5 mg/46 mg을 초과해서는 안 된다.
이 약은 신장 투석 중에 있는 말기 신장질환 환자에 대해서는 연구되지 않았다. 이러한 환자군에서는 이 약의 사용을 피해야 한다.
11. 간장애 환자
경증(Child‑Pugh 5 ‑ 6) 및 중등증(Child‑Pugh 7 ‑ 9) 간장애 환자에서 펜터민에 대한 노출은 건강한 지원자에 비해 더 높았다. 이 약 성분인 토피라메이트에 대한 노출은 경증 및 중등증의 간장애 환자와 건강한 지원자 간에 유사했다.
경증의 간장애가 있는 환자에게는 용량 조절이 필요하지 않다. 중등증의 간장애가 있는 환자의 경우 1일 1회 7.5 mg/46 mg을 초과해서는 안 된다.
이 약은 중증 간장애 환자(Child‑Pugh score 10 ‑ 15)에 대해서는 연구되지 않았다. 이러한 환자군에서는 이 약의 사용을 피해야 한다.
12. 과량투여시의 처치
이 약을 과다 복용하는 경우, 과다 복용한지 얼마 되지 않았다면 즉시 위세척이나 구토를 통해 위를 비워야 한다. 환자의 임상 징후와 증상에 따라 적절한 보조요법(supportive treatment)이 제공되어야 한다.
급성 펜터민 과다 복용은 불안, 진전, 과다반사, 빠른 호흡, 착란, 공격성, 환각 그리고 공황 상태를 일으키는 경우가 있다. 피로와 우울증은 대개 중추 자극(central stimulation)에 의한다. 심혈관 영향에는 부정맥, 고혈압 또는 저혈압, 순환허탈 (circulatory collapse) 등이 있다. 위장 증상으로는 오심, 구토, 설사, 복부경련이 있다. 치명적인 중독은 보통 경련과 혼수상태로 이어진다. 식욕억제를 동반하는 만성 중독 증상은 심각한 피부병, 현저한 불면증, 과민성, 과행동증 및 성격 변화를 포함한다. 만성 중독의 심각한 징후는 정신증이며, 종종 임상적으로 정신분열증과 구별할 수 없다.
급성 펜터민 중독의 관리는 주로 증상을 관찰하는 것이며 세척과 바르비탈계 약물을 통한 진정이 포함된다. 뇨의 산성화는 펜터민 배설을 증가시킨다. 이 약의 과량 투여로 인한 급격하고 심각한 고혈압에는 가능한 신속히 펜톨아민 정맥주사하는 것이 추천된다.
토피라메이트 과다 복용으로 심각한 대사성산증이 초래되었다. 다른 징후와 증상으로는 경련, 졸림, 언어 장애, 시야흐림, 복시, 정신기능 저하, 졸음증, 협조이상, 혼미, 저혈압, 복통, 초조, 어지러움, 우울증이 있다. 대부분의 경우 임상 결과는 심각하지 않았지만, 다량의 토피라메이트를 포함한 여러 종의 약물 과다 복용 후 사망이 보고되었다. 96 ~ 110 그램의 토피라메이트를 복용한 환자는 20 ~ 24시간 지속되는 혼수상태로 입원한 다음 3 ~ 4일 후에 완전히 회복되었다.
약용탄은 시험관내에서 토피라메이트를 흡착하는 것으로 나타났다. 혈액 투석은 체내에서 토피라메이트를 제거하는 효과적인 수단이다.
13. 보관 및 취급상의 주의사항
1) 어린이의 손이 닿지 않는 곳에 보관하도록 주의한다.
2) 다른 용기에 바꾸어 넣는 것은 사고원인이 되거나 품질 유지 면에서 바람직하지 않으므로 주의한다.
14. 전문가를 위한 정보
1) 약력학적 정보
암페타민의 전형적인 작용에는 중추 신경계 자극 및 혈압 상승이 포함된다. 타키필락시스(급성 내성)와 내성은 이러한 현상이 발견된 이 부류의 모든 약물에서 입증되었다.
심장 전기생리학
무작위, 이중 맹검, 위약 대조 및 활성 대조군(목시플록사신 400 mg) 및 평행군/교차, QT/QTc 연구를 통해 QTc 간격에 대한 이 약의 영향을 평가했다. 총 54명의 건강한 피험자에게 정상 상태에서 이 약 7.5 mg/46 mg을 투여한 다음 정상 상태에서 이 약 22.5 mg/138 mg으로 적정했다. 이 약 22.5 mg/138 mg [이 약 7.5 mg/46 mg의 펜터민과 토피라메이트 최대 농도(Cmax)에 대해 각각 4배 및 3배 높은 최대치료용량] 는 QTc의 기저치로부터의 변화를 측정했을 때 심장 재분극에 영향을 미치지 않았다.
사구체여과율(GFR)
건강한 비만 남성 및 여성은 이 약(1~3일 3.75mg/23mg, 4~6일 7.5mg/46mg, 7~9일 11.25mg/69mg, 10~28일 15mg/92mg)을 4주간 복용했다. 피험자의 사구체여과율은 이오헥솔 청소율을 통해 평가되었다. 평균적으로 이 약 치료기간동안 사구체여과율은 감소하였고 이 약 복용 중단 후 4주 이내에 기저치로 회복되었다.
<BR><SPAN style="FONT‑SIZE: 11pt">2) 약물동력학적 정보</SPAN>
펜터민
이 약 단회 용량 15 mg/92mg을 경구 투여한 결과, 평균 혈장 펜터민 최대 농도(Cmax), Cmax 까지의 시간(Tmax), 투약시간부터 최종 혈중농도 정량시간 t까지의 혈중농도‑시간곡선하면적(AUC0‑t), 투약시간부터 무한시간까지의 혈중농도‑시간곡선하면적 (AUC0‑∞)은 각각 49.1ng/mL, 6시간, 1990ng▪hr/mL 및 2000ng▪hr/mL이다. 이 약 15mg/92mg에 대한 고지방 식이는 펜터민 약물동력학에 영향을 미치지 않는다. 펜터민 약물동력학은 대략 이 약 3.75mg/23mg에서 펜터민 15mg/토피라메이트 100mg까지 용량에 비례한다. 펜터민/토피라메이트 15/100mg 복합제 캡슐을 정상 상태에서 투여할 때, AUC 및 Cmax의 평균 펜터민 축적 비는 모두 약 2.5이다.
토피라메이트
이 약 단회 용량 15mg/92mg을 경구 투여한 결과, 평균 혈장 토피라메이트의 Cmax 및 Tmax, AUC0‑t 및 AUC0‑∞는 각각 1020 ng/mL, 9시간, 61600 ng▪hr/mL 및 68000 ng▪hr/mL이었다. 이 약 15mg/92mg에 대한 고지방 식이는 토피라메이트 약물동력학에 영향을 미치지 않는다. 토피라메이트 약물동력학은 대략 이 약 3.75mg/23mg에서 펜터민 15mg/토피라메이트 100mg까지 용량에 비례한다. 펜터민 15mg/토피라메이트 100mg 투여량의 복합제 캡슐을 정상 상태에서 투여할 때, AUC 및 Cmax의 평균 토피라메이트 축적 비는 모두 약 4.0이다.
분포
펜터민
펜터민은 17.5%의 혈장 단백질과 결합한다. 예상되는 펜터민 분포 용적(Vd/F)은 개체군 약물동력학적 분석 결과 348 L이다.
토피라메이트
토피라메이트는 0.5~250μg/mL 혈중농도범위에서 15‑41% 혈장 단백질과 결합한다. 혈중 토피라메이트가 증가함에 따라 결합 분획(fraction bound)이 감소했다. 개체군 약물동력학적 분석을 통해 추정된 토피라메이트 Vc/F(중앙 구획의 용적) 및 Vp/F(말초 구획의 용적)는 각각 50.8 L 및 13.1 L이다.
신진 대사 및 배설
펜터민
펜터민은 두 가지 대사경로, 즉 방향족 고리(aromatic ring)에 p‑hydroxylation과 지방족 측쇄(aliphatic side chain)에 N‑oxidation을 가지고 있다. Cytochrome P450(CYP) 3A4는 주로 펜터민을 대사하지만 광범위한 신진 대사를 보이지는 않는다. Monoamine oxidase(MAO)‑A와 MAO‑B는 펜터민을 대사하지 않는다. 단독 투여 시 뇨에서 대사되지 않은 70 ~ 80%의 펜터민이 존재한다. 평균 펜터민의 말단 반감기는 약 20시간이다. 개체군 약물동력학적 분석을 통해 예상되는 펜터민 경구 청소율(CL/F)는 8.79L/h이다.
토피라메이트
토피라메이트는 광범위한 신진 대사를 보이지 않는다. 6개의 토피라메이트 대사물질(하이드록실화(hydroxylation), 가수분해(hydrolysis) 및 글루크론산화 (glucuronidation)을 통한)이 존재하며, 그 중 어떤 것도 투여된 용량의 5% 이상을 차지하지 않는다. 단독투여시 뇨에서 대사되지 않은 70% 용량의 토피라메이트가 존재한다. 평균 토피라메이트의 말단 반감기는 약 65시간이다. 개체군 약물동력학적 분석을 통해 예상되는 토피라메이트 CL/F는 1.17L/h이다.
특정환자집단
신장애
정상적인 신장 기능을 가진 건강한 지원자와 비교하여 만성 신장애 정도가 다양한 환자에서 이 약 15 mg/92 mg의 약물동력학을 평가하기 위해 단회투여, 공개 임상시험이 수행되었다. 이 연구에는 크레아티닌 청소율을 기준으로 경증(50 이상 80 mL/min 미만), 중등증(30 이상 50 mL/min 미만) 및 중증(30 mL/min 미만)으로 분류된 신장애 환자가 포함되어 있다. 크레아티닌 청소율은 C‑G 공식(Cockcroft‑Gault equation)에 기초하여 혈청 크레아티닌으로부터 추정하였다.
건강한 지원자와 비교했을 때, 펜터민의 AUC0‑inf는 중증, 중등증 및 경증의 신장애 환자에서 각각 91%, 45%, 22%였다. 펜터민 Cmax는 2 ~ 15% 더 높았다. 건강한 지원자와 비교했을 때, 토피라메이트 AUC0‑inf는 중증, 중등증 및 경증의 신장애 환자에서 각각 126%, 85%, 25%였다. 토피라메이트 Cmax는 6 ~ 17% 더 높았다. 펜터민 또는 토피라메이트 Cmax 또는 AUC와 크레아티닌 청소율 사이의 반비례 관계가 관찰되었다. 이 약은 신장 투석 중인 말기 신장질환 환자에서는 연구되지 않았다.
간장애
경증(Child‑Pugh score 5 ‑ 6)와 중등증(Child‑Pugh score 7 ‑ 9)의 간장애 환자에 비해 정상 간 기능을 가진 건강한 지원자에서의 이 약 15 mg/92 mg의 약물동력학을 평가하기 위해 단회투여, 공개 임상시험이 수행되었다. 경증 및 중등증의 간장애 환자에서, 펜터민 AUC는 건강한 지원자와 비교하여 37% 및 60% 더 높았다. 토피라메이트의 약물동력학은 건강한 지원자와 비교할 때 경증 및 중등증의 간장애 환자에서는 영향을 받지 않는다. 이 약은 중증 간장애 환자(Child‑Pugh score 10‑15)에서는 연구되지 않았다.
약물상호작용
약물상호작용의 시험관내 평가
펜터민
펜터민은 CYP 동종 효소 CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 및 CYP3A4의 억제제가 아니며 모노아민 산화 효소의 억제제도 아니다. 펜터민은 CYP1A2, CYP2B6 및 CYP3A4의 유도 인자가 아니다. 펜터민은 P‑당단백질(P‑glycoprotein) 기질이 아니다.
토피라메이트
토피라메이트는 CYP 동종 효소 CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2D6, CYP2E1 및 CYP3A4/5의 억제제가 아니다. 그러나, 토피라메이트는 CYP2C19의 경도(mild) 억제제이다. 토피라메이트는 CYP3A4의 경도(mild) 유도 인자이다. 토피라메이트는 P‑당단백질(P‑glycoprotein) 기질이 아니다.
다른 약물에 미치는 펜터민/토피라메이트의 영향
표 6. 펜터민/토피라메이트가 병용투여 약물의 약물동력학에 미치는 영향
표 7. 병용투여 약물이 펜터민/토피라메이트의 약물동력학에 미치는 영향
항경련제
간질 환자를 대상으로 한 대조 임상 약물동력학 연구에서 토피라메이트와 표준 항경련제(AED) 간의 잠재적인 상호 작용을 평가했다. 이러한 상호 작용이 평균 혈장 AUC에 미치는 영향을 표 7에 요약했다.
표 8에서 두 번째 열(AED 농도)은 토피라메이트가 추가될 때 첫 번째 열에 나열된 항경련제(AED) 농도에 어떤 현상이 발생하는지를 설명한다. 세 번째 열(토피라메이트 농도)은 토피라메이트 단독 투여 시 실험 환경에서의 토피라메이트 농도를 첫 번째 열에 기재된 약물의 병용투여가 어떻게 변화시키는지를 설명한다.
표 8. 토피라메이트와의 항경련제(AED) 상호 작용 요약
단회 투여 연구에서 토피라메이트의 병용투여로 혈장농도곡선 아래 혈청 디곡신 면적(AUC)이 12% 감소되었다. 이러한 관찰결과에 대한 임상적 관련성은 확립되지 않았다.
히드로클로로티아지드(HCTZ)
건강한 지원자를 대상으로 실시한 약물 상호작용 연구는 정상 상태에서의 히드로클로로티아지드(25 mg 매24시간마다) 및 토피라메이트(96 mg 매12시간마다) 단독 및 병용 투여 후 약물동력학을 평가하였다.
이 연구 결과 HCTZ을 추가하였을 때 토피라메이트의 Cmax가 27%, AUC가 29% 증가되었다. 이러한 변화의 임상적 유의성은 알려져 있지 않다. 정상 상태에서의 HCTZ의 약물동력학은 토피라메이트의 병용으로 유의한 영향을 받지 않았다. 토피라메이트와 HCTZ 투여 후 혈청 칼륨의 저하가 관찰되었으며 두 약물을 병용 투여했을 때 더 크게 나타났다.
피오글리타존
건강한 자원자를 대상으로 한 약물상호작용 연구에서 7일 동안 토피라메이트(96mg 1일 2회)와 피오글리타존(30mg 1일)을 단독 및 병용 투여 후 정상상태에서의 약물동력학을 평가하였다. 피오글리타존의 AUCt,ss가 15% 저하되었으며 Cmax,ss에는 변화가 관찰되지 않았다. 이 결과는 통계적으로 유의하지 않았다. 활성 수산화‑대사체의 Cmax,ss와 AUCt,ss는 각각 13%, 16% 저하되었으며, 활성 keto‑대사체의 Cmax,ss와 AUCt,ss는 60% 저하되었다. 이 결과의 임상적 유의성은 알려져 있지 않다. 피오글리타존 치료를 받는 환자에 이 약을 투여하거나 이 약을 투여받는 환자에 피오글리타존을 투여할 때 당뇨가 적절히 조절되고 있는지 모니터하는데 주의한다.
글리부리드
제2형 당뇨병 환자를 대상으로 한 약물상호작용 연구에서 글리부리드 단독(5mg/day) 및 토피라메이트와 병용(150mg/day)시의 정상상태 약물동력학을 평가하였다. 토피라메이트 투여 중 Cmax는 22% 감소하였고 글리부리드 AUC24가 25% 감소하였다. 또한, 활성 대사체 4‑trans‑히드록시글리부리드(M1) 및 3‑cis‑히드록시글리부리드(M2)의 전신노출은 각각 13% 및 15%까지 저하되었고, Cmax는 각각 18%와 25% 감소했다. 토피라메이트의 정상상태 약물동력학은 글리부리드의 병용투여에 영향을 받지 않았다.
리튬
토피라메이트 200 mg/day 투여 중 환자에서 리튬의 약물동력학은 영향을 받지 않았다. 그러나 토피라메이트를 600 mg/day까지 투여한 후 리튬의 전신 노출이 증가하는 것으로 나타났다(Cmax는 27%, AUC는 26%). 고농도의 토피라메이트와 병용 투여시 리튬 수치를 모니터링해야 한다.
할로페리돌
할로페리돌(5mg) 단회 투여의 약물동력학은 13명의 건강한 성인(남성 6명, 여성 7명)에서 토피라메이트(12시간마다 100mg)의 반복 투여에 의한 영향을 받지 않았다.
아미트립틸린
토피라메이트 200 mg/day를 투여받은 일반인 18명(남성 9명, 여성 9명)에서 아미트립틸린(하루 25mg)의 AUC와 Cmax가 12% 증가했다. 일부 피험자에서 토피라메이트를 복용 시 아미트립틸린 농도가 크게 증가할 수 있으며, 아미트립틸린 용량 조절은 혈청 수치가 아닌 환자의 임상 반응에 따라 이루어져야 한다.
수마트립탄
24명의 건강한 지원자(남성 14명, 여성 10명)에서 토피라메이트(12시간마다 100mg)의 반복 투여는 단회 투여의 수마트립탄의 경구(100mg) 또는 피하 투여(6mg)의 약물동력학에 영향을 미치지 않았다.
리스페리돈
토피라메이트를 100, 250, 400mg/day 로 용량을 증가하여 병용투여시 리스페리돈의 전신 노출이 감소되었다(250, 400mg/day에서 각각 정상상태 AUC의 16%, 33% 감소). 9‑히드록시히스페리돈 수치의 변화는 관찰되지 않았다. 리스페리돈과 토피라메이트 400 mg/day 병용투여 시 토피라메이트의 Cmax가 14% 증가하였고, AUC12가 12% 증가했다. 리스페리돈과 9‑히드록시히스페리돈 또는 토피라메이트의 전신 노출에 대해 임상적으로 유의한 변화는 없었으므로, 이 상호작용은 임상적으로 유의하지 않을 것으로 예상된다.
프로프라놀롤
34명의 건강한 지원자(남성 17명, 여성 17명)에 대한 토피라메이트(200mg/day) 반복 투여는 일일 160mg 용량의 프로프라놀롤 약물동력학에 영향을 미치지 않았다. 39명의 지원자(남성 27명, 여성 12명)에서 160mg/day의 프로프라놀롤 복용량은 200mg/day 용량의 토피라메이트 노출에 아무런 영향을 미치지 않았다.
디히드로에르고타민
24명의 건강한 지원자(남성 12명, 여성 12명)에서 토피라메이트(200mg/day)의 반복 투여는 1mg 피하 투여량의 디히드로에르고타민 약물동력학에 영향을 미치지 않았다. 유사하게, 디히드로에르고타민의 1 mg 피하 투여량은 동일한 연구에서 토피라메이트의 200 mg/day를 투여했을 때의 약물동력학에 영향을 미치지 않았다.
딜티아젬
토피라메이트(150mg/day)와 딜티아젬(240mg)을 병용투여 시, 딜티아젬 Cmax가 10% 감소하고 AUC가 25% 감소했으며, des‑acetyl diltiazem의 AUC는 18%, Cmax가 27% 감소하고, N‑desmethyl diltiazem에는 영향을 미치지 않았다. 토피라메이트와 딜티아젬의 병용투여는 결과적으로 토피라메이트의 Cmax를 16% 증가시키고, AUC12를 19% 증가시킨다.
벤라팍신
건강한 지원자에서 토피라메이트(150mg/day)를 반복투여 시 벤라팍신 또는 O‑desmethyl 벤라팍신의 약물동력학에 영향을 미치지 않았다. 벤라팍신(150 mg 서방제제)의 반복 투여는 토피라메이트의 약물동력학에 영향을 미치지 않았다.
3) 독성시험 정보
⓵ 발암성, 변이원성, 수태능이상
펜터민/토피라메이트
펜터민/토피라메이트 복합제로 발암성, 변이원성 또는 수태능이상을 평가한 동물 실험은 없었다. 다음 데이터는 이 약의 두 가지 주성분인 펜터민 또는 토피라메이트로 개별적으로 수행한 연구 결과를 바탕으로 한다.
펜터민
펜터민은 Ames 박테리아 변이원성 시험, Chinese 햄스터 폐 세포로 시행한 염색체 이상 분석 또는 생체내 소핵 분석에서 대사 활성화 또는 대사 증진이 없는 변이원성 또는 염색체성 물질이 아니다.
랫드에게 3, 10, 30 mg/kg/day의 펜터민을 2년간 경구 투여하였다. AUC 노출에 근거하여, 이 약 15mg/92mg의 최대 권장 투여량의 약 11~15배인 최고 용량의 펜터민(30 mg/kg)에서 발암성에 대한 증거는 없었다.
수태능이상에 대한 잠재력을 결정하기 위해 펜터민으로 수행한 동물실험은 없었다.
토피라메이트
토피라메이트는 일련의 시험관내/생체내 분석 시험에서 유전 독성 잠재력을 보이지 않았다. 토피라메이트는 Ames 시험이나 시험관내 마우스 림프종 시험에서 돌연변이성을 갖지 않았다. 그것은 시험관내에서 랫드의 간세포에서 부정기 DNA 합성(unscheduled DNA synthesis)을 증가시키지 않았으며, 시험관 내 인간 림프구나 생체 내 랫드의 골수에서 염색체 이상을 증가시키지 않았다.
21개월 동안 식사를 통해 토피라메이트(20, 75 및 300 mg/kg)를 투여한 마우스에서 방광 종양의 증가가 관찰되었다. 300 mg/kg을 투여 받은 암컷과 수컷에서 통계적으로 유의한 방광 종양 발생률의 상승은 주로 마우스에 조직 형태학적으로 고유한 평활근 종양의 증가로 인한 것이다. 300 mg/kg을 투여 받은 마우스의 혈장 노출은 이 약 15 mg/92 mg의 인체최대권장용량인 토피라메이트 단일요법을 받은 환자에서 측정한 정상 상태 노출의 약 2 ~ 4배이었다. 인체 발암성에 대한 이러한 발견의 관련성은 아직은 불확실하다. 최대 120 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 약 4 ~ 10배)까지의 용량으로 2년간 토피라메이트를 경구 투여한 결과 랫드에서 발암성의 증거는 발견되지 않았다.
최대 100mg/kg 또는 AUC에 근거한 이 약의 남성 및 여성에 대한 인체최대권장용량의 약 4 ~ 8배 노출 시 랫드에서 암수컷의 수태능에 영향이 없었다.
⓶ 생식발생독성시험
토피라메이트
이 약 성분인 토피라메이트는 여러 동물 종에서의 임상적으로 적절한 용량에서 최기형성을 비롯한 발생 독성을 유발한다.
기관형성기에 임신한 마우스에게 20, 100, 500 mg/kg으로 경구 투여했을 때, 모든 투여량에서 태아 기형 초기 두개안면결손) 발생률이 증가했다. 이 연구에서 토피라메이트 저용량(20 mg/kg)은 이 약의 15 mg/92 mg에서 mg/m2기준으로 토피라메이트의 인체최대권장용량의 약 2배이다. 태아의 체중과 골격골형성은 모체 체중 증가의 감소와 함께 500 mg/kg으로 감소되었다.
랫드 연구 (경구 투여량 20, 100, 500 mg/kg 또는 0.2, 2.5, 30, 400 mg/kg)에서 사지기형 (손발가락결손증 (ectrodactyly), 사지단소증 (micromelia), 무지증 (amelia))의 빈도는 임신 중 기관형성기간 동안 400 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량 34배) 이상으로 치료한 암컷의 자손사이에서 증가했다. 배아독성(태아 체중 감소, 구조적 변화의 발생률 증가)은 20 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량 2배) 정도의 낮은 용량에서 관찰되었다. 모체 독성에 대한 임상적 징후는 400 mg/kg 이상에서 나타나고, 모체의 체중 증가는 100 mg/kg 혹은 그 용량 이상으로 치료하는 동안 감소했다.
토끼 연구(기관형성기간 동안 경구로 20, 60, 180 mg/kg 또는 10, 35, 120 mg/kg)에서는 배/태아 사망률이 35 mg/kg(AUC 추정치에 근거한 인체최대권장용량의 2배)이상에서 증가였고, 최기형성의 영향(주로 늑골 및 척추 기형)이 120 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 6배)에서 관찰되었다. 모체 독성(체중 증가의 감소, 임상적 증후 및/또는 사망률)의 증거가 35 mg/kg 이상에서 나타났다.
임신 후반기와 수유기(0.2, 4, 20, 100 mg/kg 또는 2, 20, 200 mg/kg)의 암컷 랫드에 투여했을 때, 자손은 200 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 16배)에서 생존력이 감소하고 신체 발달이 지연되었으며, 2 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 2배) 이상에서 이유기 전 및/또는 후에 체중 증가가 감소하는 것으로 나타났다. 모체 독성(체중 증가의 감소, 임상 징후)은 100 mg/kg 혹은 그 이상에서 나타났다.
산후 성분(기관형성 중 0.2, 2.5, 30, 400 mg/kg)을 갖은 랫드의 배아/태아 발생 연구에서, 자손은 400 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 34배)에서 신체 발달이 지연되었고, 30 mg/kg(AUC 추정치에 근거한 이 약의 인체최대권장용량의 2배)이상에서 체중 증가의 지속적인 감소가 나타났다.
⓷ 임상시험 정보
감소된 칼로리 섭취 및 늘어난 신체 활동과 관련하여 체중 감량에 대한 이 약의 효과는 2개의 무작위배정, 이중맹검, 위약 대조 임상시험에서 비만 환자군(시험 1)와 비만 혹은 2가지 이상의 유의한 동반질환을 갖는 과체중 환자군(시험 2)에서 연구되었다. 두 임상시험 모두 4주간의 적정 기간이후 52주간의 치료 기간이 있었다. 치료 1년 후(56 주)에 측정된 2가지의 공동 1차 유효성 결과지표는 1) 기저치로부터의 체중 감소 백분율과 2) 기저치로부터 최소한 5%의 체중 감소 달성으로 정의되는 치료에의 반응이었다.
시험 1에서 비만 환자군(BMI 35kg/m2이상)는 1년 동안 2:1:2의 비율로 514명은 위약을, 241명은 이 약 3.75mg/23mg을, 512명은 이 약 15mg/92mg을 무작위로 처방 받았다. 환자는 18 ~ 71세(평균 연령 43세)로 구성되었으며, 83%가 여성이었다. 약 80%는 백인, 18%는 아프리카 계 미국인, 15%는 히스패닉/라틴계였다. 임상시험 시작 당시 환자의 평균 체중과 BMI는 각각 116kg과 42kg/m2였다. 제2형 당뇨 환자는 시험 1 참여에서 제외되었다. 이 임상시험 동안에 모든 환자에게 칼로리 섭취량이 약 500 kcal/day 감소하도록 균형 잡힌 저칼로리 식이 요법을 권장했으며, 영양 및 생활습관의 변화에 관한 상담을 제공했다.
시험 2에서 과체중 및 비만 환자군은 1년 동안 2:1:2의 비율로 994명은 위약을, 498명은 이 약 7.5mg/46mg을, 995명은 이 약 15mg/92mg을 무작위로 처방받았다. 임상시험에 적합한 환자는 27kg/m2 이상 45kg/m2 이하의 BMI(제2형 당뇨 환자의 경우 BMI에 대한 하한 값은 없음)와 다음의 비만관련 합병증에서 2가지 이상의 조건을 충족해야 했다.
• 고혈압(140/90 mmHg 이상 또는 당뇨인 경우 130/85 mmHg 이상) 또는 2가지 이상의 혈압강하제가 필요한 환자
• 200‑400 mg/dL 이상의 트리글리세리드 또는 2개 이상의 지질 저하 약물로 치료를 받고 있는 환자
• 공복 혈당 상승(100 mg/dL 이상) 또는 당뇨병;
• 허리 둘레: 남성 102cm 이상, 여성 88cm 이상
환자는 19 ~ 71세(평균 연령 51세)로 구성되었으며, 70%가 여성이었다. 약 86%는 백인, 12%는 아프리카 계 미국인, 13%는 히스패닉/라틴계였다. 임상시험 시작 당시 환자의 평균 체중과 BMI는 각각 103kg과 36.6kg/m2였다. 임상시험 시작 시 환자의 약 절반(53%)이 고혈압을 앓았다. 연구 시작 당시 제2형 당뇨 환자는 388명 (16%)이었다. 이 연구 동안에 모든 환자에게 칼로리 섭취량이 약 500 kcal/day 감소하도록 균형 잡힌 저칼로리 식이 요법을 권장했으며, 환자에게 영양 및 생활습관의 변화에 관한 상담을 제공했다.
무작위 배정된 환자 중 시험 1에서 40%, 시험 2에서 31%에 해당하는 상당수의 피험자가 56주 전에 연구를 중단하였다.
표 9은 시험 1과 시험 2에서 1년간의 체중 감량 결과를 나타낸다. 이 약 치료 1년 후, 모든 용량에서 위약에 비해 통계적으로 유의한 체중 감량을 초래했다 (표 9, 그림 1 및 2). 위약군 대비 이 약에 무작위 배정된 환자들의 통계적으로 상당히 유의한 비율에서 5% 및 10%의 체중감량을 달성하였다.
표 9. 시험 1과 시험 2에서 1년간의 체중 감량
그림 1. 시험 1 체중 변화 비율 그림 2. 시험 2 체중 변화 비율
시험 1과 시험 2의 비만과 관련된 심혈관, 대사 및 인체 계측 위험 요소의 변화는 표 10와 표 11에 나타내었다.
이 약을 사용한 1년간의 치료로 심박수를 제외하고 비만과 관련된 여러 위험 요소에서 위약보다 상대적으로 호전이 있었다.
표 10. 시험 1(비만)에서 1년간의 치료 후 위험 인자에 대한 기저치로부터의 최소 제곱근 평균† 변화(Least‑Squares (LS) Mean Change) 및 위약군과의 치료 결과 차이
시험 2에서 치료한 제2형 당뇨가 있는 388명의 대상자 중 기저치 대비 HbA1c 감소(6.8%)는 위약군에서는 0.1% 감소한 반면, 이 약 7.5 mg/46 mg 군 및 15 mg/92 mg군에서는 각각 0.4%씩 감소했다.